Abstract
Virtual screening with high-performance computers is a powerful and cost-effective technique in drug discovery. A chemical database is searched to find candidate compounds firmly bound to a target protein, judging from the binding poses and/or binding scores. The severe acute respiratory syndrome coronavirus 2 (SARS-Cov-2) infectious disease has spread worldwide for the last three years, causing severe slumps in economic and social activities. SARS-Cov-2 has two viral proteases: 3-chymotrypsin-like (3CL) and papain-like (PL) protease. While approved drugs have already been released for the 3CL protease, no approved agent is available for PL protease. In this work, we carried out in silico screening for the PL protease inhibitors, combining docking simulation and molecular mechanics calculation. Docking simulations were applied to 8,820 molecules in a chemical database of approved and investigational compounds. Based on the binding poses generated by the docking simulations, molecular mechanics calculations were performed to optimize the binding structures and to obtain the binding scores. Based on the binding scores, 57 compounds were selected for in vitro assay of the inhibitory activity. Five inhibitory compounds were identified from the in vitro measurement. The predicted binding structures of the identified five compounds were examined, and the significant interaction between the individual compound and the protease catalytic site was clarified. This work demonstrates that computational virtual screening by combining docking simulation with molecular mechanics calculation is effective for searching candidate compounds in drug discovery.
Introduction
In silico screening is a fundamental technique in the initial stage of drug discovery and development.1) The recent progress of computer facilities enables us to efficiently find candidate compounds for a target protein from a chemical database. Since virtual screening is a low-cost and efficient approach, the technique can be applied swiftly to many targets.2,3) The performance of in silico screening depends on the accuracy of identifying hit compounds from a chemical library. In our previous study,4) we attempted a two-step approach combining docking simulation and molecular mechanics (MM) calculation. In docking simulation, the individual compound in a chemical database is bound to the target protein. The docking score indicates the compatibility of a generated docking pose with the target site. Therefore, the docking score is usually used to rank the compounds for picking up candidates from the database. Many docking simulation programs have already been released,5–7) and each program has an advantage in prediction accuracy for specific categories of compounds. Further, some programs generate adequate binding poses, while others give reliable docking scores. Hence, it is safe to generate multiple binding poses in the stage of a docking simulation. The docking score should be re-evaluated by a more accurate computational method. The MM calculation can make the binding score feasible for reasonably selecting the potent candidate compounds.4)
Severe acute respiratory syndrome coronavirus 2 (SARS-Cov-2) outbroke the 2019 pandemic of coronavirus infectious disease. The virus has been rapidly spreading worldwide, which caused millions of deaths, anxiety in public health, and depression in socioeconomic activities. Effective SARS-Cov-2 drugs were urgently required,8,9) and approved antiviral inhibitors were investigated to be released as drug repositioning agents.10,11) Remdesivir was first approved as an antiviral drug for SARS-Cov-2 in Japan.12) Remdesivir aims at viral polymerase. Molnupiravir is a ribonucleoside-based oral prodrug also targeting viral polymerase.13) Besides drug repositioning, a novel class of inhibitors has been developed for targeting viral proteins of SARS-Cov-2. Ensitrelvir is an approved, orally administered drug that blocks the enzymatic activity of 3-chymotrypsin-like (3CL) protease.14) 3CL protease, called as main protease, functionalizes viral replication by cleaving the precursor polyproteins. Ensitrelvir was discovered via a virtual screening followed by biological screening, and the subsequent optimization was achieved through the structure-based drug design. Nirmatrelvir is another oral protease inhibitor targeting 3CL protease.15) Nirmatrelvir also exhibited a fine pharmacokinetic profile in vivo for oral dosing. Despite the release of several antiviral drugs aiming at polymerase and 3CL protease, no approved inhibitor was produced for Papain-like (PL) protease. Chemotherapy with multiple classes of drugs is effective for treating infectious diseases caused by mutant viruses. Therefore, the agents blocking the PL protease are still important.
PL protease is essentially required for viral polyprotein processing and for post-translational modifications. PL protease preferentially cleaves the amino-terminal ubiquitin-like domain of interferon-stimulated gene 15 protein (ISG15), as suggested by the crystal structure of the PL-ISG15 complex.16) An inhibitor for PL protease, GRL-0617, was obtained by a structural optimization from a hit compound found in an in vitro screening with a diverse chemical library.17) Naphthalene-based inhibitors were expected to be effective in halting SARS-Cov-2 PL protease activity as expected from the SARS-Cov inhibition.18) Modifying the naphthalene-based inhibitor improved the compound potency to 0.6 µM.19) A chemical design connecting multiple shallow binding sites on the protease surface found that substituting thiophene phenyl for naphthalene enhanced inhibitory activity to be 0.4 µM.20) An X-ray crystal analysis clarified the complex structure of SARS-Cov-2 PL protease and GRL-0617.21) Other kinds of inhibitory compounds were also identified by screening with the fluorescence resonance energy transfer (FRET) technique.21)
Besides GRL-0617 derivatives, many screening works have been carried out for the last three years. Three compounds, YM155, cryptotanshinone, and tanshinone I, were identified to inhibit SARS-Cov-2 PL protease from a chemical screening.22) An anticancer drug candidate, YM155, showed potent antiviral activity with the cell-based assay at the sub-micro molar level. Another high-throughput screening identified acriflavine (ACF) as a potent PL protease inhibitor.23) A study identified six inhibitory compounds using a collection of 40 metal-chelating chemicals. One was a hydrazone-based compound, and the other five were thiosemicarbazone-based ones.24) Another work identified piperidine-based inhibitors from a drug repurposing library of 11804 compounds.25)
In this work, a computer screening was carried out to find inhibitory agents for the SARS-Cov-2 PL protease using a chemical database of approved and investigational compounds. Docking simulations initially predicted the binding poses at the active site. The docking software has been widely employed for predicting the binding pose of low molecular weight compounds to a target protein. Since the docking scores of docking programs were insufficient in accuracy,26) the binding pose and its score need to be improved to select the candidate compounds reliably. Then, the predicted binding pose was optimized by the energy minimization with MM calculation. The binding score was evaluated by the MM calculation associated with the optimized binding pose. The MM calculation is helpful for analyzing intermolecular interactions such as compound-enzyme inhibition27,28) and antibody-antigen recognition.29,30)
The compounds selected by the computer screening were examined in an experimental assay. From the calculation, 57 compounds were purchased to examine inhibitory activity against the PL protease. First, the inhibitory potency was monitored at a constant concentration of 50 µM. Five compounds were picked up for the assay with different concentrations. Second, the IC50 values were determined with the compound solutions in serial dilution. The optimized binding structures were closely examined for the five compounds to obtain a hint for designing potent scaffolds.
Experimental
Calculation ConditionIn silico screening was performed by combining docking simulation with AutoDock Vina5) and MM calculation with Orientation.31) The docking simulation generated the predicted binding poses of individual compounds to a target protein. AutoDock Vina is frequently employed in chemical screening because the software program is applicable to a large-scale database. The calculation model for the PL protease was built from the crystal structure, PDB#: 6wuu.32) A peptide inhibitor was bound to the enzymatic active site in the 6wuu structure. Then, there appeared a space that a screening compound could be bound to. Five binding poses were generated for a compound. A search box of 38 Å × 38 Å × 48 Å was set at the cavity near Cys111 of the PL protease. Six residues, Trp106, Asn109, Cys111, Leu162, Cys270, and His272, were assigned to be flexible residues because these are located near the enzymatic active center of the PL protease.
The MM calculation optimized the binding pose and estimated the binding score. The atom pair-wise force field with Morse-type potential function was employed to estimate the binding score between a compound and the target protein. The parallel computation with a super-computer system effectively processed the predicted binding poses. The optimization of the binding pose was executed by the steepest descent method, in which energy-minimizing calculation was repeated for 50000 cycles without setting the positional constraint for any atom. The Amber ff14SB force field33) was applied to describe the intra-molecular interaction, and the Morse-type potential function31) was used to describe the inter-molecular interaction. The binding affinity of a compound was predicted from the inter-molecular interaction. The binding score was re-evaluated by including the solvent effect for the optimized binding structure.4)
Procedure for Chemical ScreeningCandidate compounds for experimental assay were searched from the DrugBank database, which consisted of 8820 approved drugs and clinically investigational chemicals. Compounds were stored in sdf format in the chemical library of the DrugBank database. The files were converted into pdbqt format to make inputs for AutoDock Vina. The atom coordinate of the PL protease was prepared with the pdbqt format, in which rigid and flexible residues are separated into two files. Docking simulations were performed for all 8820 compounds, targeting the active site of SARS-Cov-2 PL protease, and five binding poses were generated for each compound. MM calculations were performed to optimize the binding poses generated by AutoDock Vina. Based on the binding score obtained from the MM calculation, the top 200 binding structures were extracted from the predicted binding poses. The same compounds with different binding poses were occasionally included in the 200 extracted structures. All the 200 extracted structures were visually examined individually in terms of the viewpoints of whether a compound well fitted the deep hollow sites at the binding pocket and whether no large hydrophobic groups were markedly exposed to the solvent. Finally, 57 compounds were selected as the candidates for experimental assay of inhibitory activity.
Protein Expression and PurificationA recombinant PL protease was expressed with pET50b plasmid vector in which Nus-tag was conjugated at the N-terminal side. An Escherichia coli competent cell, Rosetta 2 (DE3) pLysS, was transformed with the plasmid vector, and the cell was cultured with 1.5 L Luria–Bertani medium at 37 °C. The protein expression was induced by adding 0.2 mM IPTG at an optical density (OD) 600 value of 0.6. After adding IPTG, the medium was incubated at 20 °C overnight. Then, the cultured cells were harvested by centrifugation at 5000 × g. The cells were re-suspended in 40 mL of buffer solution of 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF). The cell membrane was disrupted by ultrasonication. The Nus-tag-conjugated PL protease was purified by Co-affinity chromatography using a starting buffer of 50 mM Tris–HCl, 150 mM NaCl, pH 8.0 with 10 mM imidazole, and an elution buffer of 50 mM Tris–HCl, 150 mM NaCl, pH 8.0 with 500 mM imidazole. The imidazole was removed by dialysis with 1 L buffer solution of 50 mM Tris–HCl pH 8.0, 150 mM NaCl. The Nus-tag conjugation was cleaved by incubating human rhinovirus 3C (HRV-3C) protease at 4 °C for 24 h. Ni-affinity chromatography was performed to remove the Nus-tag, HRV-3C protease, and remaining noncleaved protein. The target protein was collected from the flow-through fractions. The PL protease was further purified by anion exchange chromatography with a starting buffer of 50 mM 2-(N-morpholino) ethanesulfonic acid (MES) pH 6.5 with 25 mM NaCl and an elution buffer of 50 mM MES pH 6.5 with 1000 mM NaCl. The protein was finally purified by gel filtration with a running buffer of 20 mM Tris–HCl at pH 7.2, 150 mM NaCl, and 1 mM dithiothreitol (DTT).
Measurement of Inhibitory ActivityThe inhibitory activity was measured by the fluorescence resonance energy transfer (FRET) assay. A fluorogenic peptide, Z-RLRGG-AMC, was utilized as a substrate for the hydrolysis by PL protease.34,35) The fluorescence intensity was monitored with a microplate reader with wavelengths of 360 nm for excitation and 485 nm for emission with a bandwidth of 20 nm. The hydrolysis reaction was performed with a buffer of 50 mM Tris–HCl pH 7.2, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA) at 25 °C, containing 0.1 nM PL protease, 4 µM fluorogenic substrate, and the individual compound. The fluorescence from the reaction mixture was monitored every 60 s for 50 min. The IC50 value was determined by plotting the initial reaction rates of the measurements at different compound concentrations in the two-fold serial dilution from 64 µM.
Results
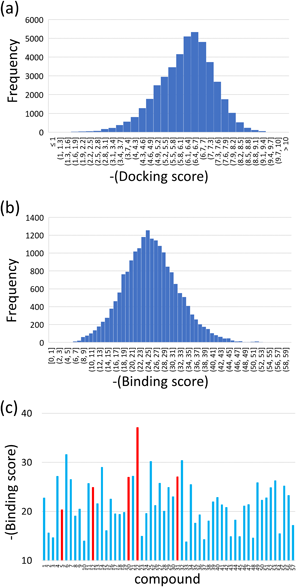
In Silico Search for PL InhibitorsThe 8820 compounds in the chemical database were bound to the SARS-Cov-2 PL protease by the docking simulations. Five binding poses were predicted for each compound, and then 44100 binding poses and docking scores were obtained by the docking simulation. The docking scores range from −10.9 to −0.3 (Fig. 1(a)). The low docking score indicated a high binding affinity to the target. The population of the docking scores had a single peak at −6.5, and the histogram showed an asymmetric distribution around the peak. One-tenth of the docking scores were lower than −7.5.
MM calculations were performed for the top 20000 predicted binding poses. The binding structure was optimized for every pose, and the binding score was also obtained by the MM calculations. The MM binding scores exhibited a symmetric distribution (Fig. 1(b)). The distribution was broad, ranging from −58.4 to −0.7. A single peak appeared at −24.7. The binding score was re-evaluated for the top 2000 optimized binding structures, scaling the score by a factor depending on the area ratio in contact with solvent waters and the target protein. When many water molecules were located around a compound, the binding score was reduced for modifying the over-estimation of the water-mediated hydrophilic interaction.4)
The top 200 structures in the MM binding score were picked up, and their chemical structures and binding poses were visually inspected. We closely examined whether a compound well fitted the deep hollow sites at the binding pocket. Compounds with excessively high hydrophobicity were excluded despite the chemicals in the approved and investigational drug database. Multiple binding poses of a compound were ranked in the top 200 structures. The binding structure with the best score was selected for the compounds in such a case. Finally, we selected 57 candidate compounds for purchase and experimental assay. The re-evaluated MM binding scores were broadly distributed even for the selected 57 candidates (Fig. 1(c)). More than half of the compounds exhibited better scores than −20. The chemical structures of the 57 compounds are listed in Supplementary Table S1.
Enzymatic Inhibitory Assay of Candidate CompoundsThe inhibitory activities of the 57 compounds were measured by an in vitro enzymatic assay. The FRET measurement was performed for all 57 compounds at a concentration of 50 µM in the first step. A fluorescence profile without any compound was referred to as a blank. Monitoring the reduction of the fluorescence intensity, we picked up the compounds for the assay in the second step. The final concentration of each compound was changed from 1 to 64 µM in a two-fold dilution series.
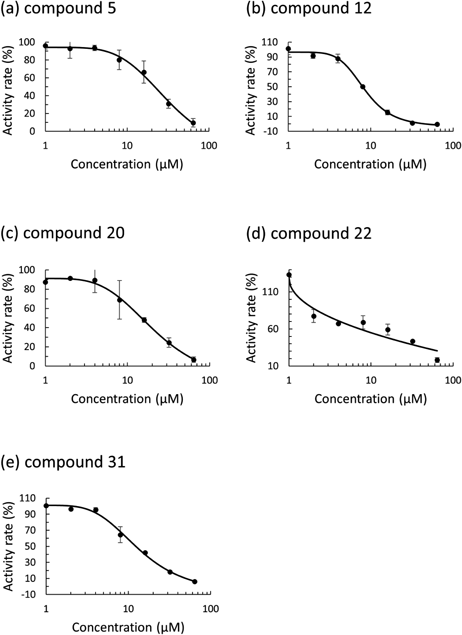
Based on the assay with serial dilution in compound concentration, we identified five compounds showing inhibitory activity for the PL protease. The numbers of the identified compounds were 5, 12, 20, 22, 31 in Supplementary Table S1. The FRET measurement was carried out twice. The enzymatic activity rate was plotted against the compound concentrations, and then the IC50 value was obtained for the five compounds (Fig. 2). The computed docking scores and binding scores of the five identified compounds were shown in Supplementary Table S2.
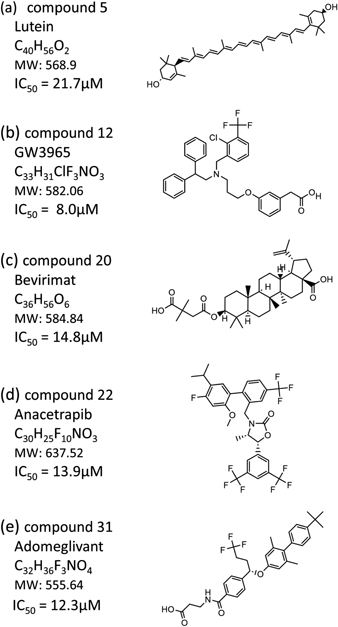
The IC50 values of the identified compounds are shown with their chemical structures (Fig. 3). Compound 5 is Lutein, an oxygen-containing carotenoid that is taken as nutritional supplementation for treating dietary shortage or imbalance. The chemical backbone of compound 5 is a long-chain carbon hydride containing many unsaturated bonds. Both sides of the long carbon chain are terminated with cyclohexanols. The IC50 value is the highest among the five identified compounds. Compound 12, GW3965, is a potent and selective agonist for liver X receptor (LXR). Compound 12 has EC50 values of 190 and 30 nM in a cell-based assay for hLXRα and hLXRβ, respectively. A diphenyl-ethyl is connected to a central N atom in compound 12. A trifluoromethyl-attached chloro-benzyl is linked to the N atom. Further, a phenylacetic acid moiety is bound to the N atom via an alkyl linker. Compound 12 exhibits the lowest IC50 value among the five compounds.
Compounds 20, 22, and 31 are Bevirimat, Anacetrapib, and Adomeglivant. These three compounds show moderate inhibitory activities of 12–15 µM in IC50. Compound 20 has a chemical scaffold of pentacyclic triterpenoid, in which a dimethyl-succinic acid is connected to a betulinic acid by an ester linkage. Compound 20 is a potent human immunodeficiency virus type 1 (HIV-1) drug candidate. The target is the HIV-1 Gag capsid precursor, in which compound 20 blocks the protease processing of the precursor polyprotein and hinders the maturation of capsid protein. Compound 22 is an investigational compound inhibiting cholesteryl ester transfer protein (CETP) for treating hypercholesterolaemia. Compound 22 reduces the transfer of cholesteryl ester from high density lipoprotein (HDL) to low density lipoprotein (LDL), which leads to an increase in HDL cholesterol and a decrease in LDL cholesterol in serum. Compound 22 is abundant in fluorine. A bis(trifluoromethyl)-phenyl and a trifluoromethyl-phenyl are connected via an oxazolidine backbone. A fluoro-methoxy-propan-phenyl is attached to the bis(trifluoromethyl)-phenyl. Compound 31 is a potent and selective allosteric antagonist of glucagon receptor (GluR) for type 2 diabetes mellitus therapy. A butyl-phenyl-connected dimethyl-phenoxy is linked to a trifluorobutyl-benzoic moiety. An amino-propanoic acid is bound to the benzoic moiety by an amide linkage.
Predicted Binding Poses of the Identified CompoundsThe binding structures optimized by the MM calculations were depicted for the identified five compounds (Fig. 4). Since PL protease is a cysteine protease, the catalytic Cys residue is located at the binding pocket of the enzyme. There are marked hollow spaces at the PL protease binding pocket. Then, we labeled three major hollow spaces as regions 1, 2, and 3 for an explanation. Region 1 is a hollow space near Cys111 and Leu162. Cys111 is the central catalytic residue of this enzyme. Region 2 is positioned near Asp164 and Arg166. Region 2 accepts a polar moiety of compounds, which enhances the binding affinity by establishing hydrogen bonds between protease and inhibitors. Region 3 is a space near Pro248, Tyr264, and Tyr 268. These residues at region 3 make hydrophobic contacts with compounds. The CH–π and π–π interactions with the aromatic residues enhance the binding affinity of inhibitors. Regions 1, 2, and 3 were noted as R1, R2, and R3 in the surface drawing of the binding structures, respectively. Schematic illustrations of the inter-molecular interactions were also depicted for the five compounds (Supplementary Fig. S1). In the docking simulation, the search box was set at the enzymatic active center. However, many compounds were docked to R2 and R3 since the space at the active center was considerably narrow.
The binding score obtained by MM calculation is an index for the strength of intermolecular interaction between a compound and the target PL protease. The binding score can be divided into the contributions of the respective atoms because the MM force field is the atom pair-wise potential function. The binding score can also be separated into the hydrophobic and hydrophilic interactions based on the atom types. Hence, the hydrophobic and hydrophilic contributions of the individual atoms to the binding score are schematically illustrated for the respective compounds (Supplementary Fig. S2).
Compound 5 contains two cyclohexanol moieties at both sides of the unsaturated alkyl chain. One cyclohexanol is firmly accommodated at R1, while R2 and R3 are unoccupied (Fig. 4(a)). An extended linear conformation is a characteristic chemical structure of compound 5, which results in a broad compound-protein contact. Leu162 makes a hydrophobic interaction with the alkyl chain of compound 5. The other cyclohexanol contacts with Trp106 through a CH–π interaction (Supplementary Fig. S1(a)). Despite the above strong interactions, many areas of compound 5 are exposed to solvent. Hence, the binding score of compound 5 is not high compared to other chemicals.
Compound 12 fits nicely into the groove of the PL binding pocket (Fig. 4(b)), in which chemical groups occupy all the R1, R2, and R3. Four aromatic rings are expanded from the central N atom in compound 12. The phenylacetic acid moiety is positioned at R1, in which the terminal carboxy group makes hydrogen bonds with the main-chain atoms of Gln269 and Gly271. The aromatic ring of the chloro-benzyl group makes an NH-π interaction with Arg166, and the trifluoromethyl firmly contacts with Thr301 at R2 (Supplementary Fig. S1(b)). Two phenyls are located at R3, in which the two phenyls make a hydrophobic contact with Pro248. Strong π–π interactions are observed with Tyr264 and Tyr268. These π–π interactions significantly stabilize the compound accommodation.
Compound 20 is characterized by a dimethyl-succinic acid connected to a betulinic acid. The carboxy group of the succinic acid occupies R2 and forms strong hydrogen bonds with Arg166 and Asp164. Tyr264 and Tyr273 make firm CH–π contacts with the betulinic acid skeleton. A hydrophobic contact with Leu162 makes the compound binding to R1 robust (Supplementary Fig. S1(c)). Despite these strong interactions, much of the compound surface is exposed to solvent. The R3 is unoccupied by the compound.
The chemical shape of compound 22 is well-compatible with the groove at the binding pocket. The compound moieties occupy all the R1, R2, and R3. R1 houses the bis(trifluoromethyl)-phenyl moiety. One trifluoromethyl fits nicely into the deep inside of R1, while the other is exposed to solvent. Another trifluoromethyl-phenyl moiety is located at R2. The trifluoromethyl is oriented to the inside of R2. An oxazolidine connect the bis(trifluoromethyl)-phenyl to the mono(trifluoromethyl)-phenyl. The oxazolidine makes a hydrogen bond with Tyr273, forming a CH–π interaction with Tyr264 (Supplementary Fig. S1(d)). The fluoro-methoxy-propan-phenyl moiety is positioned at R3. The phenyl ring makes a π–π interaction with Tyr268. The methoxy group faces the solvent, which helps stabilize the compound accommodation. The binding score is markedly high since compound 22 forms many interactions with the PL protease.
Compound 31 is characterized by a connection of two phenyl rings. The terminal butyl-phenyl is completely sticking out from the binding pocket. While the contribution of the two phenyl rings is marginal, other parts of compound 31 are well complementary with the groove of the PL protease. The trifluoromethyl group is positioned at R1, making hydrophobic contacts with Leu162 and Tyr273. The terminal carboxy group is positioned at R2, forming a strong hydrogen bond with Arg166. The phenyl in the midst of compound 31 makes two π–π interactions with Tyr264 and Tyr273 (Supplementary Fig. S1(e)).
Discussion
Because of the global pandemic of SARS-Cov-2 infectious disease, dozens of compounds were identified as PL protease inhibitors. GRL-0617 is a well-known inhibitor of PL protease.16,17) GRL-0617 has a naphthalene scaffold linked with methyl-aniline via amide linkage, and many modifications of GRL-0617 were attempted in previous studies.18–21) A derivative of the naphthalene-based inhibitor marked a compound potency of 0.6 µM.19) Much effort has been devoted to the search for inhibitory compounds other than GRL-0617.22–24) Acriflavine, an antiseptic for treating infected wounds, was identified to block the enzymatic activity of SARS-Cov-2 PL protease.23) Acriflavine is composed of an acridine backbone, in which the central C atom of anthracene is replaced by N. Therefore, planar polycyclic aromatic rings such as naphthalene and anthracene are typical scaffolds for PL protease inhibitors. The five compounds identified in this work bear neither naphthalene nor anthracene. Accordingly, the compounds can be feasible for chemical bases to develop SARS-Cov-2 PL inhibitors other than the previously reported potent scaffolds.
Some screening studies have been conducted using a chemical database of approved drugs because of the urgent requirement for effective chemotherapeutic agents for SARS-Cov-2 infectious disease.22,25) In this work, we selected 57 compounds for experimental assay based on the results of a computational screening. Some compounds were already tested in inhibitory activity or proposed as SARS-Cov-2 inhibitors. As long as PL protease inhibitor, a few compounds in the 57 candidates were pointed out in the previous studies. Compound 4, Petesicatib, was previously evaluated with other compounds,36) and no inhibitory activity was observed. Compound 30, Sacubitril, was proposed as one of the 44 candidates for experimental assay from a molecular docking calculation.37) Compound 48, Enasidenib, showed a high binding score with 147 other candidates in a docking simulation.38) Therefore, previous screening studies on the approved drugs were limited to the PL protease inhibitors. The present study is one of the reports that clarify the inhibitory activity with the approved and investigational compounds. In contrast to PL protease, many of the selected 57 compounds were tested for inhibitory activity against 3CL protease (Supplementary Table S3). Since the 3CL protease is an excellent therapeutic target for SARS-Cov-2 infectious diseases,39) the compound potencies were evaluated by many studies with in vitro screening, cell-based antiviral assay, and computational approaches.
In this work, an in silico screening was carried out by combining docking simulation and subsequent MM calculation. From the DrugBank database, 57 compounds were selected as candidates for experimental assay. Five compounds were found to have inhibitory activity for PL protease. The five inhibitory compounds showed good binding scores among the 57 candidates (Fig. 1(c)). However, some other compounds indicated high binding scores despite no inhibitory potency in the in vitro assay. Three compounds, 6, 25, and 32, showed good binding scores, and the absolute values of these three are beyond 30. The predicted binding poses of these three compounds were examined to clarify the reason for the high scores (Supplementary Fig. S3).
In compound 6, Terfenadine, (Supplementary Fig. S3(a)), the aromatic rings expanded from the central piperidine occupy all the R1, R2, and R3 regions. The tert-butyl phenyl fits well into R1. The tert-butyl makes a hydrophobic contact with Leu162. The hydroxy group near the tert-butyl phenyl has a hydrogen bond with the main-chain carbonyl oxygen. One phenyl is positioned at R2, but no hydrogen bonds are formed with Asp164 and Arg166. Another phenyl is located at R3, which establishes π–π interactions with Tyr264 and Tyr268 and makes a hydrophobic contact with Pro248. Compound 6 shows fine shape compatibility with the binding pocket as long as the computational prediction. In compound 25, Lomitapide, (Supplementary Fig. S3(b)), R1 and R3 areas are occupied by the aromatic rings, while R2 is vacant. A trifluoro-benzene is positioned at the deep inside of R1. The phenyl ring connecting the trifluoro-benzene makes a CH–π interaction with Leu162. Further, a fluorene at the opposite side establishes strong π-π contacts with Tyr264 and Tyr268. However, the piperidine ring at the center position and the trifluoro-ethyl linked to the fluorene scarcely fit in the binding pocket. The exposure of these hydrophobic moieties to solvent is disadvantageous for the stability of compound binding. In compound 32, Ebastine, (Supplementary Fig. S3(c)), R1, R2, and R3 are occupied by chemical moieties appropriately. A tert-butyl phenyl is located at R1, in which the tert-butyl makes a hydrophobic contact with Leu162. The center propyl-piperidine moiety occupies R2, and the piperidine N atom makes a hydrogen bond with Arg166. Biphenyl moiety is positioned at R3, where two aromatic rings establish strong π–π and CH–π interactions with Tyr264, Tyr268, and Pro248. The binding pocket of the PL protease is composed of many aromatic residues. Every compound of 6, 25, and 32 contains considerably large hydrophobic substitutes. Since those substitutes can establish strong interactions with those aromatic residues, the binding scores in the MM calculations are opted to be better than other compounds. A penalty function for the unfavorable contact with solvent may improve the accuracy of the binding score. Hence, an exact solvation effect estimation is needed to enhance the computational accuracy. As well as compound 6, the solubility of compound 32 seems insufficient due to the high hydrophobicity.
The compounds containing naphthalene moiety are potent inhibitors for SARS-Cov-2 PL protease. Many phenyl-based substitutes are bound to the naphthalene 8-th position via amide linkage.20) A crystallographic study suggested that the naphthalene-based inhibitors occupied R2 and R3 in Fig. 4.40) The naphthalene was placed at R3, and the phenyl substitutes were positioned at R2. Pro247, Pro248, Tyr264, and Tyr268 held the naphthalene moiety. Therefore, R3 is essential for linking the compounds to the binding pocket. The substitutes connecting to the naphthalene should be complementary to R2 in shape. If a small functional group is connected to the R2 occupying substitutes and the group is extended to the direction of R1, the compound potency will be increased significantly. Among the five compounds identified in this work, compounds 12 and 22 occupy all R1, R2, and R3 regions. Therefore, both compounds are suitable for the starting chemical backbones to develop potent RNase H inhibitors. Since the chemical groups positioned at R3 are a phenyl and substitutes-connected phenyl, a larger moiety can be allocated at R3. Modification by increasing the interaction at R3 will enhance the inhibitory activities of compounds 12 and 22.
Conclusion
Computational screening for inhibitors against the SARS-Cov-2 PL protease was carried out, combining docking simulation with MM calculation. A chemical search for the PL inhibitors was performed with a database of approved drugs and agents used in clinical trials. The docking simulation generated the binding poses of the individual compound in the database to the catalytic site of PL protease. The binding poses were optimized by the MM calculations. The MM calculations also predicted the binding scores of the respective compounds to the target site. The binding scores ranged from −58.4 to −0.7, with a symmetric single peak at −24.7. The binding score was re-evaluated for the top 2000 optimized binding structures, including the influence of water molecules. Visually inspecting the chemical structures of the top 200 compounds, we selected 57 compounds for experimental assay. The enzymatic inhibitory activity was evaluated by the FRET method. The assay was performed for all 57 compounds at a constant concentration of 50 µM. Based on the assay at a constant concentration, five compounds were identified to inhibit the PL protease enzymatic action. The FRET measurements with a dilution series of compound concentrations gave the IC50 values for all five compounds. Among the five compounds, a preclinical compound, GW3965, exhibited the lowest IC50 value of 8.0 µM. The predicted binding structures of the identified compounds were examined in terms of interactions with the target site. This work demonstrated that the combinatorial virtual screening effectively found inhibitor candidates from a chemical database.
Acknowledgments
Calculations of this work were performed at OBCX super-computer system of Information Technology Center of the University of Tokyo under the support of HPCI System Research Project (Project ID: hp210017 and hp220103). A part of the calculation was executed at Research Center for Computational Science, Okazaki, Japan (Project: 22-IMS-C001). This study was supported by a Grant for Scientific Research C (21K07050) and Transformative Research A (23H04462) from Japan Society for the Promotion of Science.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
Supplementary materials contain a list of the candidate compounds utilized for the experimental assay, computed docking score and binding score, a list of the previously reported assay, interaction diagrams of the inhibitory compounds in the binding pocket, and docking poses of the three compounds with fine binding scores.
References
- 1) Shaker B., Ahmad S., Lee J., Jung C., Na D., Comput. Biol. Med., 137, 104851 (2021).
- 2) Nakamura Y., Suganami A., Fukuda M., Hasan M.-K., Yokochi T., Takatori A., Satoh S., Hoshino T., Tamura Y., Nakagawara A., Cancer Medicine, 3, 25–35 (2014).
- 3) Yanagita H., Yamamoto N., Fuji H., Liu X., Ogata M., Yokota M., Takaku H., Hasegawa H., Odagiri T., Tashiro M., Hoshino T., ACS Chem. Biol., 7, 552–562 (2012).
- 4) Miwa K., Guo Y., Hata M., Yamamoto N., Hoshino T., Chem. Pharm. Bull., 71, 360–367 (2023).
- 5) Trott O., Olson A. J., J. Comput. Chem., 31, 455–461 (2010).
- 6) Eberhardt J., Santos-Martins D., Tillack A. F., Forli S., J. Chem. Inf. Model., 61, 3891–3898 (2021).
- 7) Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., Banks J. L., J. Med. Chem., 47, 1750–1759 (2004).
- 8) Vandyck K., Deval J., Curr. Opin. Virol., 49, 36–40 (2021).
- 9) Herlah B., Hoivi A., Jamšek L., Valjave K., Yamamoto N., Hoshino T., Kranjc K., Perdi A., Pharmaceuticals, 15, 539 (2022).
- 10) Yamamoto N., Matsuyama S., Hoshino T., Yamamoto N. Nelfinavir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro. bioRxiv, 2020, doi: https://doi.org/10.1101/2020.04.06.026476
- 11) Shiraki K., Daikoku T., Pharm. Therapeutics, 209, 107512 (2020).
- 12) Enoki Y., Igarashi Y., Watabe Y., Honma K., Suzuki Y., Hayashi Y., Hiraoka K., Taguchi K., Matsumoto K., J. Glob. Antimicrob. Resist., 24, 81–82 (2021).
- 13) Syed Y. Y., Drugs, 82, 455–460 (2022).
- 14) Unoh Y., Uehara S., Nakahara K., et al., J. Med. Chem., 65, 6499–6512 (2022).
- 15) Xie Y., Choi T., Al-Aly Z., JAMA Intern. Med., 183, 554–564 (2023).
- 16) Shin D., Mukherjee R., Grewe D., et al., Nature (London), 587, 657–662 (2020).
- 17) Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B. S., Johnson M. E., Baker S. C., Ghosh A. K., Mesecar A. D., Proc. Natl. Acad. Sci. U.S.A., 105, 16119–16124 (2008).
- 18) Freitas B. T., Durie I. A., Murray J., Longo J. E., Miller H. C., Crich D., Hogan R. J., Tripp R. A., Pegan S. D., ACS Infect. Dis., 6, 2099–2109 (2020).
- 19) Shan H., Liu J., Shen J., Dai J., Xu G., Lu K., Han C., Wang Y., Xu X., Tong Y., Xiang H., Ai Z., Zhuang G., Hu J., Zhang Z., Li Y., Pan L., Tan L., Cell Chem. Biol., 28, 855–865.e9 (2021).
- 20) Shen Z., Ratia K., Cooper L., Kong D., Lee H., Kwon Y., Li Y., Alqarni S., Huang F., Dubrovskyi O., Rong L., Thatcher G. R. J., Xiong R., J. Med. Chem., 65, 2940–2955 (2022).
- 21) Ma C., Sacco M. D., Xia Z., Lambrinidis G., Townsend J. A., Hu Y., Meng X., Szeto T., Ba M., Zhang X., Gongora M., Zhang F., Marty M. T., Xiang Y., Kolocouris A., Chen Y., Wang J., ACS Cent. Sci., 7, 1245–1260 (2021).
- 22) Zhao Y., Du X., Duan Y., et al., Protein Cell., 12, 877–888 (2021).
- 23) Napolitano V., Dabrowska A., Schorpp K., et al., Cell Chem. Biol., 29, 774–784.e8 (2022).
- 24) Ewert W., Günther S., Miglioli F., Falke S., Reinke P. Y. A., Niebling S., Günther C., Han H., Srinivasan V., Brognaro H., Lieske J., Lorenzen K., Garcia-Alai M. M., Betzel C., Carcelli M., Hinrichs W., Rogolino D., Meents A., Front. Chem., 10, 832431 (2022).
- 25) Calleja D. J., Kuchel N., Lu B. G. C., et al., Front. Chem., 10, 861209 (2022).
- 26) Zev S., Raz K., Schwartz R., Tarabeh R., Gupta P. K., Major D. T., J. Chem. Inf. Model., 61, 2957–2966 (2021).
- 27) Kamo T., Kuroda K., Kondo S., Hayashi U., Fudo S., Yoneda T., Takaya A., Nukaga M., Hoshino T., Chem. Pharm. Bull., 69, 1179–1183 (2021).
- 28) Qu L., Fudo S., Matsuzaki K., Hoshino T., Chem. Pharm. Bull., 67, 959–965 (2019).
- 29) Qu L., Qiao X., Qi F., Nishida N., Hoshino T., J. Chem. Inf. Model., 61, 2396–2406 (2021).
- 30) Qiao X., Qu L., Guo Y., Hoshino T., J. Phys. Chem., 125, 11374–11385 (2021).
- 31) Fuji H., Qi F., Qu L., Takaesu Y., Hoshino T., Chem. Pharm. Bull., 65, 461–468 (2017).
- 32) Rut W., Lv Z., Zmudzinski M., Patchett S., Nayak D., Snipas S. J., El Oualid F., Huang T. T., Bekes M., Drag M., Olsen S. K., Sci. Adv., 6, eabd4596 (2020).
- 33) Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., Simmerling C., J. Chem. Theory Comput., 11, 3696–3713 (2015).
- 34) Daczkowski C. M., Dzimianski J. V., Clasman J. R., Goodwin O., Andrew D., Mesecar A. D., Pegan S. D., J. Mol. Biol., 429, 1661–1683 (2017).
- 35) Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A. D., Baker S. C., J. Virol., 79, 15189–15198 (2005).
- 36) Cho C. C., Li S. G., Lalonde T. J., Yang K. S., Yu G., Qiao Y., Xu S., Ray Liu W., ChemMedChem, 17, e202100455 (2022).
- 37) Sencanski M., Perovic V., Milicevic J., Todorovic T., Prodanovic R., Veljkovic V., Paessler S., Glisic S., ChemistryOpen, 11, e202100248 (2022).
- 38) Kouznetsova V. L., Zhang A., Tatineni M., Miller M. A., Tsigelny I. F., Peer J., 8, e9965 (2020).
- 39) Konno S., Kobayashi K., Senda M., et al., J. Med. Chem., 65, 2926–2939 (2022).
- 40) Osipiuk J., Azizi S. A., Dvorkin S., et al., Nat. Commun., 12, 743 (2021).