Abstract
This study investigates the efficacy of modified Albizia procera gum as a release-retardant polymer in Diltiazem hydrochloride (DIL) matrix tablets. Carboxymethylated Albizia procera gum (CAP) and ionically crosslinked carboxymethylated Albizia procera gum (Ca-CAP) were utilized, with Ca-CAP synthesized via crosslinking CAP with calcium ions (Ca2+) using calcium chloride (CaCl2). Fourier Transform (FT) IR analysis affirmed polymer compatibility, while differential scanning calorimetry (DSC) and X-ray diffraction (XRD) assessed thermal behavior and crystallinity, respectively. Zeta potential analysis explored surface charge and electrostatic interactions, while rheology examined flow and viscoelastic properties. Swelling and erosion kinetics provided insights into water penetration and stability. CAP’s carboxymethyl groups (–CH2–COO−) heightened divalent cation reactivity, and crosslinking with CaCl2 produced Ca-CAP through –CH2–COO− and Ca2+ interactions. Structural similarities between the polymers were revealed by FTIR, with slight differences. DSC indicated modified thermal behavior in Ca-CAP, while Zeta potential analysis showcased negative charges, with Ca-CAP exhibiting lower negativity. XRD highlighted increased crystallinity in Ca-CAP due to calcium crosslinking. Minimal impact on RBC properties was observed with both polymers compared to the positive control as water for injection (WFI). Ca-CAP exhibited improved viscosity, strength, controlled swelling, and erosion, allowing prolonged drug release compared to CAP. Stability studies confirmed consistent six-month drug release, emphasizing Ca-CAP’s potential as a stable, sustained drug delivery system over CAP. Robustness and accelerated stability tests supported these findings, underscoring the promise of Ca-CAP in controlled drug release applications.
Introduction
The field of pharmaceutical research continually strives to develop innovative drug delivery systems that enhance therapeutic outcomes while minimizing adverse effects. Among these systems, matrix tablet formulations have emerged as a promising strategy for achieving controlled drug release.1) Such formulations provide a stable platform to regulate the release of therapeutic agents, optimizing their efficacy and patient compliance. Albizia procera, sourced as an exudate from the Albizia tree belonging to the Mimosaceae family, serves as a carbohydrate polymer. It can be employed as an excipient in the production of sustained-release drug delivery systems. This choice is attributed to its notable biocompatibility and biodegradability properties.2) According to numerous research studies, it has been documented that Albizia procera gum (AP) is composed of β-(1→3)-D-galactopyranose units, along with some β-(1→6)-D-galactopyranose units, as well as α-(1→3)-L-arabinofuranose units.2) These monomeric units are characterized by the presence of hydroxyl groups on their side chains. These hydroxyl groups can be chemically modified to form carboxymethyl groups, resulting in the formation of carboxymethylated Albizia procera gum (CAP). In light of these advancements, the present study embarks on an extensive investigation into the fabrication and characterization of matrix tablet formulations, utilizing CAP3) and Ca-CAP as matrix materials in conjunction with Diltiazem hydrochloride (DIL) as a model drug. The significance of controlled drug delivery systems has been acknowledged in numerous studies. The potential of matrix tablets in prolonging drug release and maintaining therapeutic levels over an extended period, thereby enhancing patient compliance and reducing dosing frequency.4) Natural polymers gain attention in pharmaceuticals due to biocompatibility and sustainability. Researchers use carboxymethylated plant gum for sustained drug release in plant-based polymers for controlled drug delivery.5) This investigation systematically explores critical phases, revealing underlying mechanisms governing matrix tablet formulations. It extensively analyzes the polymer’s rheological behavior, vital for processing and performance, using techniques like flow curves and amplitude sweep studies. This focus on rheological properties aligns with research highlighting the correlation between polymer rheology and drug release behavior.6) Matrix tablet response to water affects drug release. Swelling, erosion, and water penetration studies reveal behavior in aqueous environments. Similar findings link polymer erosion to drug release in hydrophilic matrices.7) Interactions between polymers and the model drug are meticulously explored through Fourier Transform (FT) IR spectrum and differential scanning calorimetry (DSC) thermogram analyses. Such in-depth investigations into polymer-drug interactions which underscore the importance of understanding these interactions to predict drug release behavior.8) The study’s core intention lies in establishment of controlled release activity of the tailored matrix tablet formulations, which is ascertained by comprehensive evaluation of drug release kinetics.9) Further enriching the study, visualization of dynamic changes in tablet surface topography during dissolution using scanning electron microscopy (SEM) provides insights into dissolution kinetics, surface topography, and structural integrity of tablet.10) To ensure practical feasibility, an accelerated stability study coupled with similarity studies emphasizes the formulation’s robustness under challenging conditions.11) This investigation explores the design, development, and evaluation of controlled drug delivery systems. The objectives entail a comprehensive approach: initiating the preparation of Ca-CAP, conducting a meticulous characterization of polymers, ingredients, and the drug components, delving into the viscoelastic properties of the polymers, crafting tablets using modified polymers to analyze their unique attributes, establishing in-vitro release profiles, and scrutinizing accelerated stability for a holistic assessment of the formulations. A comprehensive in-vitro red blood cell (RBC) lysis test was conducted using CAP and Ca-CAP solutions to evaluate the substance’s capacity to induce the breakdown or destruction of red blood cells, occurring outside of a living organism. This evaluation helps determine the substance’s impact on cellular membranes and its hemolytic properties, often crucial in understanding the safety profile of various compounds or formulations. The meticulous characterization, analysis, and evaluation offered by the research contribute substantially to the field of pharmaceutical research and practice. This study’s findings hold the promise of advancing drug delivery strategies, thereby fostering enhanced therapeutic outcomes and transforming the landscape of pharmaceutical formulations.
Experimental
MaterialsDIL, Colloidal anhydrous silicon dioxide (Aerosil), and Microcrystalline cellulose (MCC, PH 102) were provided by Stadmed Pvt. Ltd., Kolkata, India. The carboxymethylated CAP (with a degree of substitution of 0.51) was obtained from the native Albizia procera gum sourced from Mizoram (Mizoram, India). Sodium hydroxide, tri-sodium phosphate dodecahydrate (TSPD), and Tri-sodium citrate (TSC) were obtained from Loba Chemie Pvt. Ltd., Mumbai, India. Calcium chloride dihydrate and analytical reagent grade methanol (99% (v/v)) were sourced from Merk Specialties Pvt. Ltd., Mumbai, India. Lactose anhydrous was from Oxford Lab Fine Chem LLP, Mumbai. Magnesium stearate (MS) was purchased from Merck Specialties Private Limited, Mumbai, India. A 0.9% w/v sodium chloride (NaCl) solution was procured from Pharma Impex Laboratories Pvt. Ltd., Kolkata, India. Other chemicals and reagents used were of analytical grade, ensuring experimental accuracy and reliability.
MethodsPreparation of Ca+2 Crosslinked CAPA 10-g portion of semi-crystalline CAP with a degree of substitution of 0.51 was sieved through a BS screen #45 and mixed with de-ionized water under stirring for 1 h, forming a slurry. A 2% (w/v) sodium hydroxide (NaOH) solution (10 mL) was gradually added to dissolve the powder, followed by the introduction of this mixture into a 5% (w/w) calcium chloride (CaCl2) aqueous solution (50 mL) with agitation. After overnight settling, the resulting mass was vacuum-filtered, the cake washed with methanol, and then dried at 60 °C for 4 h to obtain dried Ca-CAP, which was further characterized.12)
FTIR Analysis of CAP, Ca-CAP, DIL, and MixturesSamples of finely powdered CAP, Ca-CAP, DIL, and DIL with polymer mixtures were individually subjected to FTIR spectroscopy using an ATR-FTIR (Attenuated Total Reflection Fourier Transform Infrared) spectrophotometer (Alpha II, Bruker, U.K.). ATR-FTIR eliminates the requirement for KBr (potassium bromide) sample pelletization. This technique was designed to overcome the need for pelletization,13) a conventional preparation method in traditional transmission FTIR spectroscopy. To analyze solid samples, gently apply pressure to the sample against the crystal surface. The solid powdered samples underwent analysis at an optimized resolution of 4 cm−1, with 16 integrations executed to enhance the signal-to-noise ratio in the resulting spectrum.
Thermal Compatibility StudyDSC thermographs of CAP, Ca-CAP, DIL, and DIL with polymer mixtures were acquired using a DSC-4000 instrument (Perkin-Elmer, U.S.A.). The samples were hermetically sealed in aluminum pans and subjected to heating from 30 to 300 °C at a rate of 10 °C/min under a nitrogen flow. At the time of the experiment, the weight of each polymer sample was taken at 4 mg, whereas each DIL sample was taken at 3 mg.
Comparative Assessment of Zeta Potential MeasurementsZeta potential measurements were conducted utilizing a Zeta-sizer Nano ZS90 instrument (Malvern Instruments Ltd., U.K.). A 1% (w/v) dispersion of polymers was prepared at a neutral pH for the analysis. Deionized water served as the dispersion medium, and disposable zeta cells (DTS 1070) were employed for each sample at a temperature of 25 °C. The experiments were replicated thrice, and the average values were recorded. Data analysis was performed using the “Zetasizer” software version 7.03 PSS0012 - 34 EN-JP (Malvern Instruments Ltd.).
X-Ray Powder Diffraction AnalysisX-Ray diffraction (XRD) patterns were obtained for powdered polymer samples using an X-ray diffractometer (ULTIMAIII, Rigaku, Japan). The X-ray generator operated at a voltage of 40 kV and a current of 30 mA, utilizing K-β filtered Cu radiation at 1.54056 Å as the radiation source. The powdered samples were scanned over a diffraction angle (2θ) range of 5 to 80° at a scanning speed of 3°/min.
In-Vitro RBC Lysis TestThe purpose of this study is to investigate the impact of different sample solutions, specifically CAP and Ca-CAP, on red blood cells (RBCs) and their properties. The study involves a series of procedures to prepare and analyze blood samples in the presence of these sample solutions. Fresh goat blood was procured from a slaughterhouse, and 80 mL of the blood underwent preservation using a 10% (w/v) Tri-sodium citrate solution within a 0.9% (w/v) NaCl solution. This preservation method effectively prevents blood clotting and preserves the blood sample’s integrity. The preserved blood was then transferred to 1.5 mL Eppendorf tubes and subjected to centrifugation at 2000 rpm for 10 min, resulting in the separation of blood components. A dark red-colored pellet, primarily composed of RBCs, formed at the bottom, while the liquid supernatant containing plasma and other components was discarded.14) The RBC pellets underwent triple washing with freshly prepared phosphate buffer (pH 7.4) to ensure purity and eliminate impurities. Subsequently, the washed RBC pellets were suspended in phosphate buffer solution (PBS) to create a 1% RBC suspension, which was then used for the experiment. For the experiment, each well of a 96-well plate was loaded with 30 µL of the 1% RBC suspension. To this, an additional 30 µL of PB solution was added, followed by varying volumes of sample solutions to achieve a final well volume of 150–200 µL. The plate was incubated at 37 °C for 1 and 3 h, allowing the samples to interact with the RBCs. Post-incubation; the optical density of the samples was measured at 540 nm using a Multimode plate spectrophotometer. This measurement provided insights into changes in RBC properties, as indicated by alterations in optical density. To validate results, two control samples were employed: marketed water for injection as the positive control (for comparison with expected RBC behavior) and a 0.9% (w/v) NaCl solution as the negative control (to assess background effects). This experimental setup facilitated the assessment of the impact of different sample solutions on RBCs by analyzing changes in optical density, thus revealing potential effects on cellular properties.15)
Comparative Rheological AnalysisRheological analyses were performed on the polymer matrices (5% (w/v)) of both CAP and Ca-CAP, which were prepared using deionized water as the dispersion medium. The rheological experiments were conducted using a Modular Compact Rheometer (Anton Parr, MCR 102, Austria) equipped with a standard 1° cone geometry (CP-40) having a diameter of 40 mm. Each experiment was conducted at different pH levels using 5% (w/v) polymer matrices at a temperature of 25 °C. The air pressure was maintained at 6.8646 bar using a compressor unit. Two test methods were employed: dynamic rotational mode (flow curve) and oscillatory mode. For the flow curve study, the dynamic rotational mode was utilized to investigate the shear viscosity of the samples at varying shear rates. Additionally, the impact of pH on the shear viscosity of the polymeric matrices was assessed. In the amplitude sweep study, the oscillatory mode was employed to explore amplitude sweep and frequency sweep experiments. This aimed to analyze the storage and loss moduli (G′ and G″) of the polymers. The amplitude sweep was conducted at a fixed angular frequency16) (ω = 6.2831853 rad/s), where the strain (γ = 0.01–10%) was adjusted to induce structural deformation within the entangled polymer. Furthermore, the frequency sweep study was carried out to examine changes in G′ and G″ against a range of variable angular frequencies (ω = 0.1–10 rad/s). This was accomplished by utilizing the critical strain (0.3% for CAP, 2% for Ca-CAP) obtained from the amplitude sweep conducted within the linear viscoelastic (LVE) regime.17)
Preparation of Matrix Tablets: Placebo and DIL-LoadedMatrix tablets were formulated using the wet-granulation method. Ingredients were accurately weighed as per batch size. Drug, polymer, lactose, and MCC were sieved through mesh #40. Drug was mixed with polymer and other ingredients for 15 min. Dry mixture was moistened, forming cohesive mass, screened through mesh #16 for moist granules. Granules were dried at 65 °C until moisture content reached 2% (w/v). Dried granules were sieved through #20, lubricated with 0.25 to 5% (w/w) magnesium stearate and 0.1 to 0.5% (w/w) aerosol. Lubricated granules were compressed into tablets using tablet compression machine. Compression force was adjusted for tablets with 49.05 to 68.67 Newton hardness.18) To explore the swelling and erosion characteristics of the matrix, inert ingredients were used to formulate placebo matrix tablets, omitting the drug component. Both formulations, X1 and X2, were prepared with a consistent amount of polymer, with X1 containing CAP and X2 containing Ca-CAP.
For the DIL-loaded matrix tablets, the active ingredient DIL was incorporated along with CAP and Ca-CAP as polymers. The weight ratios specified in Tables 1, 2 were maintained for both placebo and DIL-loaded formulations.
Table 1. Formulation of Placebo Matrix Tablets
| Sl No. | Ingredients per tablet (mg) | Formulation code |
|---|
| X1 | X2 |
|---|
| 1. | CAP | 600 | — |
| 2. | Ca-CAP | — | 600 |
| 3. | Lactose anhydrous | 380 | 380 |
| 4. | MCC | 100 | 100 |
| 5. | Purified water | Qs* | Qs* |
| 6. | Magnesium stearate | 18 | 18 |
| 7. | Aerosil | 2 | 2 |
*Qs indicates quantity sufficient.
Table 2. Formulation of DIL Matrix Tablets
| Sl No. | Ingredients per tablet (mg) | Formulation code |
|---|
| F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 |
|---|
| 1 | DIL | 120 | 120 | 120 | 120 | 120 | 120 | 120 | 120 | 120 | 120 |
| 2 | CAP | 120 | 360 | 540 | 600 | 780 | — | — | — | — | — |
| 3 | Ca-CAP | — | — | — | — | — | 120 | 360 | 540 | 600 | 780 |
| 4 | Lactose anhydrous | 680 | 450 | 300 | 260 | 100 | 680 | 450 | 300 | 260 | 100 |
| 5 | MCC | 160 | 150 | 120 | 100 | 80 | 160 | 150 | 120 | 100 | 80 |
| 6 | Purified water | Qs* | Qs* | Qs* | Qs* | Qs* | Qs* | Qs* | Qs* | Qs* | Qs* |
| 7 | Magnesium stearate | 18 | 18 | 18 | 18 | 18 | 18 | 18 | 18 | 18 | 18 |
| 8 | Aerosil | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
*Qs indicates quantity sufficient.
The swelling analysis of placebo matrix tablets (X1, X2) was conducted in acid solution of pH 1.2, and Phosphate buffer solution of pH 6.8 using a USP II tablet dissolution test apparatus (model TDP-06P, Electrolab, India) at 37 ± 0.5 °C. Each tablet was weighed, placed in a stainless steel wire mesh basket, and immersed in 900 mL of test medium. The basket was stirred at 75 rpm. After a predetermined time, the hydrated tablet was removed, excess water was blotted, and the new weight was measured. The percentage swelling was calculated using the formula:
 | (1) |
Where W1 represents the initial weight of the tablet at time 0 and W2 is the weight of the tablet at time t after immersion in the test medium.
Erosion StudyThe erosion study was conducted following a methodology similar to the swelling study, with the only difference being the removal of the basket containing the placebo matrix tablet from the test medium (acid solution of pH 1.2, and Phosphate buffer solution of pH 6.8) at specific time intervals. The basket was then dried in a hot air oven at 70 °C until it achieved complete dryness. The percentage erosion at different time points was calculated using Equation:
 | (2) |
W1 denotes the initial weight of the tablet at the starting time (time 0), while W3 signifies the tablet’s weight at time t following its immersion in the test medium and subsequent drying.
Water Penetration Velocity in Matrix TabletsThe determination of water penetration velocity in a tablet through a swelling study provides insights into the rate at which water permeates the tablet structure, which can have implications for the tablet’s disintegration, dissolution, drug release, and overall performance. Water penetration velocity data can be used in mathematical models to predict drug release behavior under different conditions, aiding in formulation design.19)
Water penetration velocities were ascertained utilizing the following equation
 | (3) |
Here, V stands for the velocity of water penetration, ds/dt denotes the gradient of the curve illustrating the relationship between percentage swelling and the square root of time ( ), ρ symbolizes the density of the water, acid solution, and PB solution at a temperature of 310 K, and A represents the tablet’s surface area.
), ρ symbolizes the density of the water, acid solution, and PB solution at a temperature of 310 K, and A represents the tablet’s surface area.
The surface area of the caplet-shaped convex tablet can be calculated by considering its curved top surface, bottom surface, and the lateral surface connecting them.20) The determination of the surface area was accomplished using the subsequent formula:
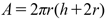 | (4) |
In this context, h signifies the thickness of the tablet, while r represents its radius, both of which were ascertained through the utilization of calipers. The radius (r) of the cylindrical shape was calculated as half of the length (length/2).
Tablet Characterization and Drug Content DeterminationWeight Uniformity AssessmentThe weight uniformity test followed the established procedure.21) From each formulation, 20 tablets were subjected to analysis using a precision electronic balance (Precisa, XB600 M-C, Switzerland). The tablets were selected randomly and weighed individually. The individual weights (m1, m2…..m20) were compared to the average weight of the tablets.
 | (5) |
The crushing strength evaluation involved testing 10 tablets from each formulation utilizing a pre-calibrated Monsanto hardness tester (Cadmach, Ahmedabad, India). The tablet to be assessed was positioned between the spindle and anvil. The required pressure for maintaining the tablet’s position was applied by turning the screw knob clockwise.22) The scale was adjusted to set the indicator at zero, and then pressure was steadily increased until the tablet fractured.
Tablet Physical Dimensions MeasurementThe physical dimensions of the tablets, including length, breadth, and thickness, were determined using a calibrated digital caliper. Measurements were taken for ten tablets from each formulation.
Friability AssessmentThe friability of 20 tablets from each formulation was evaluated utilizing the Roche friabilator, provided by Campbell Electronics, Mumbai, India. To ascertain the weight loss percentage, the friabilator was set to rotate at a speed of 25 ± 1 rpm for 4 min (equivalent to 100 revolutions). Following the process, the tablets were de-dusted and reweighed. The friability percentage was computed using the subsequent equation:
 | (6) |
Where W1 represents the initial weight of the tablets before tumbling and W2 is the weight of the matrix tablets after tumbling.
Content Uniformity for Tablets Containing DILTen tablets were weighed and crushed with a pestle in a mortar. The resulting fine powder was weighed to obtain 100 mg (equivalent to 120 mg of Diltiazem HCl) and transferred to a 250 mL conical flask containing 100 mL of a 6.8 pH phosphate buffer. The mixture was stirred for 45 min using an ultrasonic bath (sonicator). Afterward, the solution was filtered through Whatman filter paper (pore diameter 11 µm), and the drug content was determined by analyzing it using UV spectrophotometry at a wavelength (λmax) of 236 nm. The content of C22H26N2O4S, HCl (diltiazem hydrochloride) was determined by calculating the specific absorbance at 236 nm of the reference solution (RS) containing 0.0012% (w/v) of diltiazem hydrochloride in PB solution of pH 6.8.23)
In-Vitro Drug Release Study from DIL-Loaded Matrix TabletsTo examine the in-vitro drug release from matrix tablets containing DIL, a USP II tablet dissolution tester (Electrolab, TDP-06P, India) was employed with acidic (pH 1.2) and phosphate buffer (pH 6.8) solutions. The experiment was conducted in a cylindrical dissolution vessel (1000 mL) under stirring at 100 rpm. The temperature was maintained at 37 ± 0.5 °C with 750 mL of 0.1 M hydrochloric acid solution. A tablet from each formulation was randomly placed in the vessel. Throughout the experiment, hourly aliquots were drawn and replaced with an equal volume of fresh medium at the same temperature. After 2 h of dissolution in an acidic medium, 250 mL of 0.2 M tri-sodium phosphate dodecahydrate solution (previously held at 37 ± 0.5 °C) was introduced. The medium’s pH was adjusted to 6.8 ± 0.05 using a 2M sodium hydroxide solution. The dissolution process was sustained for 12 h. The collected aliquots were filtered, appropriately diluted, and subjected to spectrophotometric analysis. Drug release quantification was performed at the wavelength of maximum absorbance (λmax), found to be 236 nm for DIL in both acidic and buffer solutions.
Analysis of Drug Release Kinetics and Mechanism in in-Vitro Release ProfileThe drug release profiles were subjected to analysis using various kinetic models, including zero-order, first-order, Higuchi, and Korsmeyer–Peppas models. By comparing the obtained results, the release mechanisms of different formulations were elucidated based on the best fit to these models. The drug transport mechanism was further explored through linear regression analysis, considering the coefficient of determination values and diffusion exponent values.
To understand the drug release mechanism, the release data were fitted to exponential equations as follows:
Zero-Order Release Kinetics:
 | (7) |
Where q represents cumulative % release, K0 is the zero-order release rate constant, and t stands for time. A graph was plotted to illustrate the relationship between cumulative % drug release (Q) and time (t), and the coefficient of determination (R2) was calculated.
First-Order Release Kinetics:
 | (8) |
Here, Qt denotes the amount of drug released per unit surface area after time t, K1 is the first-order release rate constant, and Q0 is the initial amount. Another graph was plotted, depicting the relationship between the logarithm of cumulative percentage release and time. The coefficient of determination (R2) was computed to assess the fit.
Higuchi Square Root Equation:
 | (9) |
In this equation, Mt and M∞ represent cumulative amounts of drug release at time t and infinite time, respectively, while KHG signifies the Higuchi release rate constant. A plot was generated to visualize the cumulative percentage release against the square root of time, representing Higuchi release kinetics. The coefficient of determination (R2) was calculated for this representation.24)
Korsmeyer–Peppas Model:
 | (10) |
Here, Mt and M∞ represent cumulative amounts of drug release at time t and infinite time. K is a constant, and n is a diffusional release exponent that signifies the mechanism of drug release during dissolution. A graph was plotted with log [Mt/M∞] on the y-axis and log t on the x-axis. The slope of the graph was used to calculate the “n” value.25)
Scanning Electron Microscopic (SEM) StudyThe matrix tablets were collected from the dissolution chamber to study the morphological changes during the dissolution study. The surface morphology of the polymer layer was examined under SEM. Each sample was coated with gold-palladium-alloy using a fine coat ion-sputter (Hitachi, E-1010) and examined at 10.0 Kv × 100 SE and 10.0 Kv × 1.00k SE and 10.0 Kv × 500 SE with title edge of 45°. The coated samples were subsequently analyzed under a field emission Scanning Electron Microscope (Hitachi, S-3400 N).
Accelerated Stability StudyAccelerated stability studies were conducted on DIL-loaded matrix tablets from each formulation of CAP, and Ca-CAP for a duration of 6 months. The objective was to assess the stability of DIL in the respective formulations. Ten tablets from each of the formulation were placed in glass vials separately with bakelite caps, marked, and stored in a stability chamber at a temperature of 40 °C and a relative humidity of 75 ± 5% for the duration of 6 months. Upon completion of the specified duration, the samples were compared to fresh tablets in terms of drug content and dissolution profile. The similarity factor (f2) was calculated to assess the similarity between the dissolution rates of the fresh and stored tablets, utilizing the following Equation26):
 | (11) |
Where n is the number of dissolution time points, Rt is the mean percentage drug released at each time point of the reference at time t, and Tt is the mean percentage drug released at each time point of the test product at time t. The f2 comparison is widely utilized in various FDA and EMEA guidelines as a criterion to assess the similarity of in vitro dissolution profiles (Costa, 2001). An f2 value of 100 indicates identical profiles, and an average variation of 10% at all time points results in an f2 value of 50. The FDA and EMEA have set a standard range of f2 values between 50 and 100 to ensure equivalence of the two dissolution profiles.26)
Statistical Data TreatmentThe impact of different formulation parameters on drug release was assessed using ANOVA (Two-way ANOVA) at a significance level of p < 0.05.
Results
Formation of Ca-CAPCarboxymethylated CAP with a degree of substitution of 0.51 was successfully synthesized from the native Albizia procera gum. The carboxymethylation process introduced COO− groups onto the gum’s chains3) rendering them reactive with divalent cations. This property was exploited in the subsequent crosslinking step using CaCl2 to create crosslinked carboxymethylated Albizia procera gum (Ca-CAP). The successful conversion of Ca-CAP through the cross-linking of carboxymethylated gum with calcium chloride was achieved. The interaction between COO− groups on CAP chains and divalent calcium ions resulted in the formation of Ca-CAP (Fig. 1).
Characterization and Comparative StudyFT-IR Spectrum AnalysisFigures 2a and b present the Fourier transform IR (FT-IR) spectra of CAP and Ca-CAP, respectively. Despite minor variations in intensity, the positions of bands in both spectra remained largely consistent, indicating structural similarities. CAP exhibited significant absorption bands at 3292.383, 2880.317, 2360.537, 1585.374, 1459.616, 1416.322, 1321.489, and 1020.494 cm−1, which correspond to characteristic carbohydrate bands.27) Similarly, Ca-CAP displayed absorption bands at 3296.307, 2880.062, 2364.661, 1586.066, 1457.555, 1414.261, 1321.489, and 1020.371 cm−1 (Fig. 2b).
Figures 2c and d showcase the FT-IR spectra of Diltiazem HCl (DIL) and its mixtures with polymers.
DSC Thermogram AnalysisFigures 3a and b provide a comprehensive view of DSC thermograms for CAP and Ca-CAP. The data highlights distinctive thermal behavior in these polymers. CAP exhibited an exothermic event at 54.65 °C, characterized by a heat flow of 20.49 mW. In contrast, Ca-CAP displayed a slightly shifted exothermic event at 55.15 °C, accompanied by a heat flow of 21.08 mW. Both polymers exhibited endothermic peaks, with CAP showing a melting point at approximately 261.54 °C and a heat flow of 8.54 mW, while Ca-CAP demonstrated a melting peak at around 263.97 °C with a heat flow of 8.84 mW.
Figures 3c and d provide the DSC curves of the pure DIL and DIL with polymer mixtures. The endothermic peak observed at approximately 216.7 °C in both curves signifies the melting of pure DIL. This peak represents the temperature at which DIL undergoes a phase transition from a solid to a liquid state, which is a well-defined characteristic of DIL.
Zeta Potential AnalysisThe zeta potential analysis is depicted in Figs. 4A and B, yielded insightful results regarding the surface charge properties of both CAP and Ca-CAP samples. The zeta potential assessment revealed that CAP samples possessed a zeta potential value of −13.8 mV. This negative value signifies a predominance of negatively charged sites on the CAP particles’ surfaces.
XRD Pattern AnalysisThe X-ray diffractograms of CAP and Ca-CAP, as presented in Figs. 5A and B, provide valuable insights into the crystalline nature of these materials. The X-ray diffractogram of CAP reveals characteristic patterns occurring at 2θ values ranging from 15.250 to 56.500°. Significantly, the major, sharp, and intense patterns were detected at approximately 2θ values of 31.700° (756 cps), 45.450° (409 cps), and 56.5° (182 cps).
In contrast, the X-ray diffractogram of Ca-CAP exhibits characteristic patterns within a 2θ range of 17.35 to 47.1°. Notably, the major, sharp, and intense patterns observed at specific 2θ values, including 17.35° (354 cps), 18.25° (350 cps), 19.55° (391 cps), 24.7° (383 cps), 25.5° (539 cps), 28.1° (585 cps), 27.5° (503 cps), 29.4° (348 cps), 29.85° (322 cps), 33.45° (342 cps), and 47.1° (183 cps).
In-Vitro RBC Lysis TestThe study delved into the effects of CAP and Ca-CAP on RBC properties, employing optical density (OD) measurements after 1 and 3 h of incubation at 29.8 °C. The OD data, summarized in Table 3, paints a revealing picture.
Table 3. RBC Lysis Evaluation at 540 nm: 1 and 3 h Incubation
| Samples | Optical density at 540 nm |
|---|
| 1 h | 3 h |
|---|
| CAP + RBC + PBS | 0.0728 | 0.0751 | 0.0731 | 0.0751 | 0.0714 | 0.0734 | 0.0753 | 0.0763 | 0.0756 | 0.0716 |
| Ca-CAP + RBC + PBS | 0.0719 | 0.0763 | 0.0733 | 0.0755 | 0.0758 | 0.0723 | 0.0765 | 0.0738 | 0.0758 | 0.0762 |
| WFI (positive control) | 0.787 | 0.892 | 0.726 | | 0.791 | 0.897 | 0.743 | |
| Normal saline (negative control) | 0.0752 | 0.0769 | 0.0755 | | 0.0752 | 0.0769 | 0.0755 | |
| RBC suspension | 0.1687 | 0.1687 |
For CAP-treated samples after 1 h, the optical density exhibited minimal variation, signaling no discernible impact on RBC properties. This trend persisted after 3 h, indicating that CAP maintained a steady influence, effectively leaving RBCs unaffected. In the case of Ca-CAP-treated samples, the optical densities remained consistently stable, further corroborating the absence of any substantial impact on RBC properties throughout the experimental duration. However, the positive control, represented by WFI, displayed markedly higher optical densities. This discrepancy implies a substantial influence, potentially attributable to osmotic effects. Summing up, both CAP and Ca-CAP demonstrated relatively negligible effects on RBC properties when compared to the potent impact of the positive control. This suggests that these sample solutions are apt for applications related to RBCs, as they do not significantly disrupt their inherent properties.
Comparative Rheological AnalysisThe flow behavior of CAP and Ca-CAP is visually represented in Fig. 6A. These flow curves provide crucial insights into the viscosity characteristics of the two materials. Upon examining the flow curves, a notable trend becomes evident: the viscosity of the Ca-CAP matrix consistently surpasses that of CAP. This difference in viscosity profiles emphasizes the distinctive rheological behaviors exhibited by Ca-CAP and CAP. Both polymers exhibit non-Newtonian behavior characterized by pseudo-plastic flow.
Both polymers have been subjected to amplitude sweep tests, and the outcomes are presented in Fig. 6B. These parameters are observed as strain (%) is systematically varied during the course of amplitude deformation. The Ca-CAP matrix displayed a critical strain of 2%, whereas CAP showed a significantly lower critical strain of 0.3%.
During the frequency sweep analysis, the critical strain (0.3% for CAP and 2% for Ca-CAP) was maintained at a constant level within the Linear Viscoelastic (LVE) region while the angular frequency (rad/s) was systematically varied. Figure 6C illustrates the frequency sweep curves, revealing that Ca-CAP exhibited notably higher G′ values in comparison to CAP.
Figures 7a and b depict the relationship between G′ and G″ concerning angular frequency for CAP and Ca-CAP matrices. This visualization aims to comprehend the viscoelastic properties of the polymers through frequency sweep data. Comparison of R2 values (0.994 for CAP, 0.848 for Ca-CAP) and best-fit slope values (0.662 ± 0.013 for CAP, 0.254 ± 0.028 for Ca-CAP) was conducted to gain insights into the viscoelastic behavior of these polymers based on the frequency sweep data.
Table 4 displays the Trends in phase-angle (δ) for CAP and Ca-CAP matrices across various angular frequencies. Both CAP and Ca-CAP exhibit decreasing phase angles as angular frequency increases. These trends offer insights into the viscoelastic behavior of CAP and Ca-CAP matrices, with potential implications for their mechanical properties and applications. CAP generally shows higher phase angles than Ca-CAP, The rate of phase angle decrease varies between CAP and Ca-CAP, indicating differences in their viscoelastic properties.
Table 4. Phase Angle (δ) Trends of CAP and Ca-CAP Matrices across Varying Angular Frequencies
| Angular frequency (rad/sec) | Phase angle δ |
|---|
| CAP | Ca-CAP |
|---|
| 100 | 0.626666667 | 0.195859 |
| 63.1 | 0.573221757 | 0.180663 |
| 39.8 | 0.548076923 | 0.165095 |
| 25.1 | 0.537430168 | 0.15195 |
| 15.8 | 0.514102564 | 0.140986 |
| 10 | 0.497841727 | 0.132076 |
| 6.31 | 0.483606557 | 0.126432 |
| 3.98 | 0.445045045 | 0.121871 |
| 2.51 | 0.432186235 | 0.120014 |
| 1.58 | 0.448630137 | 0.119054 |
| 1 | 0.406692407 | 0.120591 |
| 0.631 | 0.358381503 | 0.125787 |
| 0.398 | 0.412520064 | 0.129687 |
| 0.251 | 0.372180451 | 0.136655 |
| 0.158 | 0.317554241 | 0.143355 |
| 0.1 | 0.300438596 | 0.152019 |
Swelling and erosion study.
Figures 8a and b showcase the swelling behavior observed in X1 and X2 placebo tablets, each containing CAP and Ca-CAP matrices, correspondingly, within acidic (pH 1.2) and buffer (pH 6.8) environments. In contrast, Figs. 8c and d present the erosion patterns evident in the CAP and Ca-CAP matrices under both acidic (pH 1.2) and buffer (pH 6.8) conditions.
Table 5 indicates that the water penetration velocity through X1 matrix tablets surpassed that of X2 matrix tablets. In addition, the visualization provided in Fig. 9 offers a comprehensive representation of the dissolution characteristics and structural integrity of X1and X2 tablets following exposure to dissolution media after 6 h.
Table 5. Water Penetration Velocity of Placebo Matrix Tablets
| Placebo matrix tablet | Water penetration velocity (cm/s) |
|---|
| In acid solution (pH 1.2) | In PB solution (pH 6.8) |
|---|
| X1 (CAP) | 1.5 × 10−3 | 1.38 × 10−3 |
| X2 (Ca-CAP) | 7 × 10−4 | 9 × 10−4 |
The physical attributes of the tablets fell within acceptable parameters. A comprehensive comparison of these physical characteristics for each formulation is provided in Table 6.
Table 6. Physical Characteristics of Tablet
| DIL-loaded tablets containing polymer | Average weight (mg) (N = 20) | Hardness (Newton/cm2) (N = 10)* | Average thickness (mm) (N = 10) * | Average length (mm) (N = 10)* | Average breadth (mm) (N = 10)* | Friability (%)(N = 20) * | Drug content (%) (N = 20) |
|---|
| CAP(F1–F10) | 1117.2–1113.04 ± 2.25% | 56.87–61.58 ±0.06 | 5.96–6.1 ±0.02 | 19.45–19.49 ± 0.011 | 8.99–9.00 ± 0.013 | 0.62–0.67 ± 0.026 | 99.08–98.98 ±0.21 |
| Ca-CAP(F1–F10) | 1114.6–1122.03 ± 2.06% | 67.27–68.74 ± 0.032 | 5.89–6.2 ± 0.01 | 19.42–19.49 ± 0.009 | 9.01–9.02 ± 0.021 | 0.56–0.61 ± 0.047 | 99.13–100.05 ± 0.19 |
* N = number of tablets tested; Mean ± standard deviation (S.D.) (n = 3).
In Table 6, it was observed that the hardness of the Ca-CAP tablet is higher than that of the CAP tablet, and the friability of the Ca-CAP tablet is lesser than that the of CAP tablet. As Ca-CAP tablet is found harder and more robust its friability is found less. This is attributed to the cross-linking of gum with calcium ions in which polymeric chains are entangled.
In-Vitro Drug Release StudyThe coefficient of correlation (R2) was obtained, and the quantity of drug dissolved in each sample was determined based on the standard calibration curve of DIL. In Figs. 10a and b, the in-vitro drug release profiles of DIL (120 mg) from tablets containing CAP and Ca-CAP in each formulation were presented. The AUC values for DIL released from CAP and Ca-CAP tablets are presented in Table 7. The drug release data were subjected to two-way ANOVA analysis to determine the significant difference in drug release between different formulations (Table 8).
Table 7.
AUC of DIL Release from Matrix Tablets
| DIL + CAP formulations | DIL + Ca-CAP formulations |
|---|
| Media | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 |
|---|
| At pH 1.2 | 269.62 | 254.08 | 249.7583 | 249.17 | 242.93 | 119.86 | 101.26 | 95.575 | 93.233 | 90.554 |
| At pH 6.8 | 1197.9 | 1179.8 | 1171.2 | 1161.9 | 1152.5 | 1585.8 | 1722.7 | 1671.354 | 1660.2 | 1629.4 |
Table 8. ANOVA Table of Cumulative Drug Release from Two Matrices
| Table analyzed |
|---|
| Two-way ANOVA | Ordinary | | | | |
| Alpha | 0.05 | | | | |
| Source of variation | % of total variation | p-Value | p-Value summary | Significant? | |
| Row factor | 26.93 | 0.0002 | *** | Yes | |
| Column factor | 3.841 | 0.738 | ns | No | |
| ANOVA table | SS | DF | MS | F (DFn, DFd) | p-Value |
| Row factor | 50774 | 12 | 4231 | F (12, 108) = 3.501 | p = 0.0002 |
| Column factor | 7242 | 9 | 804.7 | F (9, 108) = 0.6658 | p = 0.7380 |
| Residual | 130517 | 108 | 1208 | | |
| Number of missing values | 0 | | | | |
In acidic pH, AUC DIL- loaded CAP formulations (F1–F5) is higher than that of Ca-CAP formulations (F6–F10) and in pH 6.8 it shows the opposite effect.
The mechanism of drug release from hydrophilic matrix tablets is based on diffusion of the drug through, and erosion of the surrounding hydrated polymeric gel layer on the surface of the matrix. In the case of a highly soluble drug, this phenomenon may lead to an initial burst release due to the presence of the drug on the surface of the matrix tablet. In the case of the DIL-CAP tablet, it was observed that a huge amount of the drug (58–68%) was released within 1 h of dissolution and 100% of the drug was released in the 6-h experiment. The gel layer (rubbery state) grows gradually as more water permeates into the core of the matrix, thereby increasing the thickness of the gel layer and providing a diffusion barrier to drug release. Water continues to penetrate towards the core of the tablet, through the gel layer, until it has been completely eroded. The release data were evaluated by applying the equation of zero order, first order, Higuchi and Korsmeyer equation. The regression coefficient values of different release kinetic equations evaluated from the dissolution profiles of developed formulations are compared in Table 9.
Table 9. Formulation-Tailored Drug Release Kinetics Data
| Release kineticsequations | DIL-loaded CAP formulations | DIL-loaded Ca-CAP formulations |
|---|
| F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 |
|---|
| Zero order, (R2) | 0.682 | 0.718 | 0.727 | 0.731 | 0.741 | 0.872 | 0.908 | 0.922 | 0.926 | 0.923 |
| 1st order, (R2) | 0.45 | 0.464 | 0.467 | 0.468 | 0.472 | 0.584 | 0.575 | 0.580 | 0.585 | 0.584 |
| Higuchi (R2) | 0.911 | 0.933 | 0.938 | 0.911 | 0.946 | 0.985 | 0.993 | 0.985 | 0.984 | 0.983 |
| Korsmeyer–Peppas, (R2) | 0.983 | 0.978 | 0.973 | 0.979 | 0.976 | 0.949 | 0.971 | 0.945 | 0.945 | 0.947 |
| n | 0.224 | 0.271 | 0.283 | 0.282 | 0.291 | 0.381 | 0.445 | 0.464 | 0.471 | 0.463 |
Figures 11A and B reveal CAP tablet surface morphology changes during dissolution over 0–6 h, while Figs. 11C and D depict Ca-CAP tablet surface alterations at the same time points, observed via SEM.
Accelerated Stability Study of TabletsFor the stability study, the DIL-loaded tablet formulations, specifically F1, F5, F6, and F10, which exhibited the desired drug release profiles, were selected. These formulations underwent storage for a duration of six months under stress conditions, and upon thorough analysis, no significant differences in drug content or dissolution profiles were discernible (p < 0.05).
Figure 12 provides a visual representation of the release profiles for both the freshly prepared tablets and those subjected to the aging process under stressed conditions. To further assess the similarity between these two sets of data, Similarity Factors (f2) were calculated.26) Specifically, for DIL-loaded CAP tablets (F1 and F5) and Ca-CAP tablets (F6 and F10), the f2 values were determined as follows: 85.17 ± 2.64 (n = 3), 81.94 ± 2.13 (n = 3), 85.06 ± 1.83 (n = 3), and 94.30 ± 2.07 (n = 3), respectively.
Discussion
FT-IR Spectrum AnalysisWhile subtle shifts in band positions were observed, they did not signify significant structural changes between the polymers. Both CAP and Ca-CAP exhibited a broadened band between 3600–3200 cm−1, attributed to the O–H stretching of hydroxyl groups.27) The alteration appears in the FT-IR spectrum, usually as a shift in peak intensity or position within the O–H stretching region, around 3600–3200 cm−1. However, it is crucial to acknowledge that the exact nature of this shift may differ based on the particular polymer and the interaction characteristics with calcium ions. Therefore, changes in the O–H stretching band can provide an indication of calcium ion binding.28) Notably, the band for O–H stretching in Ca-CAP (Fig. 2b) exhibited heightened intensity due to crosslinking with Ca+2 ions.28) The spectra showed characteristic vibrations of C–H stretching at 2880.317 and 2880.062 cm−1.29) The appearance of bands within the 1610–1370 cm−1 range indicated the presence of carboxyl groups (–COO).30) The bending vibration of hydroxyl groups (O–H) was evident around 1459–1457 cm−1. The C=O stretching of acids was represented by the band at 1420–1418 cm−1. Furthermore, bands at 1318 and 1322 cm−1 corresponded to the scissoring of methyl groups (-CH2). The stretching frequency of >CH-O-CH was reflected by bands at 1020.49 and 1020.37 cm−1.30) The overlapping bands and consistent positions underscored the structural affinity between the two polymers.
In Fig. 2c, the distinct IR absorption bands corresponding to Diltiazem HCl were observed at specific wave numbers: 3397.57 cm−1 (N–H stretch in amine groups),31) 3034.09, 3002.50, 2939.66 cm−1 (C–H stretch), 2385.42, 1738.34 cm−1 (C=O stretch), 1675.32 cm−1 (C=C stretch), and 1604.70, 1579.76 cm−1 for C=C stretch vibrations.32) Additionally, bands at 1508.67, 1472.10, 1443.94, 1410.37, 1375.21, 1319.68, 1213.06, 1055.08 cm−1 (C–N stretch), and 1022.84 cm−1 (C–N stretch) were identified. In contrast, Fig. 2d portrays the IR absorption bands of DIL combined with polymer mixtures, exhibiting bands at 3328.91, 3055.78, 3003.55, 2916.26, 2382.93, 1742.46, 1679.58, 1606.61, 1582.14, 1538.51, 1510.19, 1473.58, 1444.51, 1412.89, 1400.34, 1217.27, 1058.08, and 1026.47 cm−1. These findings indicate a lack of significant alteration in the drug–polymer interaction.
In addition, all characteristic peaks of DIL were retained in the spectra at their respective wavenumbers, suggesting harmonious compatibility between the drug and polymer components.
DSC Thermogram AnalysisThis alteration in melting peaks suggests a modified thermal behavior in Ca-CAP. Notably, Ca-CAP exhibited a higher melting point compared to CAP, a feature attributed to the ionic crosslinking process.33)
Additionally, when DIL is combined with polymers, as shown in Fig. 3d, a similar endothermic peak emerges at a slightly lower temperature, around 215.89 °C. This shift in the melting point indicates that the presence of polymers influences the thermal behavior of DIL. It suggests that the DIL-polymer interactions may affect the energy required for DIL to transition from a solid to a liquid state. The proximity of this peak to the pure DIL melting point implies that these interactions are not significantly altering the fundamental melting behavior of DIL.34)
Zeta Potential AnalysisIn stark contrast, the zeta potential measurement for Ca-CAP samples displayed a value of −4.57 mV. The negative zeta potential of −13.8 mV for CAP is attributed to the presence of anions on the carboxymethyl groups (–OCH2–COO−) along CAP’s polymer chains. These anions contribute to the overall negative charge on CAP’s surface. A higher negative zeta potential suggests stronger electrostatic repulsion among CAP particles, contributing to improved dispersion and stability.35) In the case of Ca-CAP, despite the inherent negative charge due to carboxymethyl groups, the interactions between positively charged calcium ions (Ca2+) and carboxymethyl groups (–OCH2–COO−) on CAP’s chains led to a less negative zeta potential of −4.57 mV. The presence of calcium ions altered the zeta potential towards the positive side. However, the overall charge remained negative.
XRD Pattern AnalysisThe X-ray diffractogram analysis confirms the influence of chemical modifications (carboxymethylation and calcium crosslinking) on the crystalline properties of the polymers.36) The characteristic patterns within CAP strongly suggest the presence of crystallinity. This crystallinity is attributed to the process of carboxymethylation, which introduces structural modifications to the polymer, leading to the formation of crystalline regions. Ca-CAP demonstrates a notably higher degree of crystallinity than CAP. This increased crystallinity is attributed to the presence of calcium crosslinking, which induces more ordered and structured regions within the polymer. These differences in crystalline characteristics can have significant implications for the physical and chemical properties of CAP and Ca-CAP.
RBC Lysis TestFor CAP-treated samples after 1 h, the optical density exhibited minimal variation, signaling no discernible impact on RBC properties. This trend persisted after 3 h, indicating that CAP maintained a steady influence, effectively leaving RBCs unaffected. In the case of Ca-CAP-treated samples, the optical densities remained consistently stable, further corroborating the absence of any substantial impact on RBC properties throughout the experimental duration. However, the positive control, represented by Water for Injection (WFI), displayed markedly higher optical densities. This discrepancy implies a substantial influence, potentially attributable to osmotic effects. Summing up, both CAP and Ca-CAP demonstrated relatively negligible effects on RBC properties when compared to the potent impact of the positive control (WFI). This suggests that these sample solutions are apt for applications related to RBCs, as they do not significantly disrupt their inherent properties.
Comparative Rheological AnalysisFlow CurvesThe observed contrast in viscosity can be attributed to the unique properties of these polymers. In the case of CAP, the presence of carboxymethyl groups induces ionization, giving rise to electrostatic repulsion forces. These repulsion forces act to disentangle the polymer chains, resulting in a reduction in viscosity.37)
Conversely, the formation of Ca-CAP involves the reaction of calcium ions (Ca+2) with carboxylic groups, leading to the creation of a crosslinked structure (–OCH2COOSUP− Ca2+−OOCH2CO−).38) This crosslinking has a profound effect on the mobility of polymer chains. Specifically, it restricts their movement and significantly limits coulombic repulsion forces between them. Consequently, the chains within the Ca-CAP matrix become highly entangled, leading to a substantial increase in viscosity. This heightened viscosity, in turn, facilitates the formation of viscoelastic gels.
Amplitude SweepInitially, within the LVE, both polymers maintain their structural integrity. However, beyond a specific critical strain (%), structural deformations commence, marked by a decline in G′ that surpasses the confines of the LVE. The data are presented in Fig. 6B sheds light on these phenomena. In the context of linear viscoelasticity, it is crucial to establish the linear viscoelastic region (LVE), which denotes the range where stress exhibits a linear relationship with strain for the material under examination.39) In this regard, two critical parameters are monitored: the storage modulus G′ (Pa), which signifies the elastic response39,40) and the loss modulus G″ (Pa), which represents the viscous response. The amplitude sweep analysis conducted in this study revealed noteworthy differences in the mechanical behavior between Ca-CAP and CAP. Notably, Ca-CAP exhibited a critical strain of 2%, representing the threshold at which this polymer can withstand stress before initiating deformation. In contrast, CAP displayed a markedly lower critical strain of 0.3%, denoting its limited capacity to endure stress before deformation begins. The critical strain signifies the maximum stress threshold a material can endure without undergoing significant deformation. Beyond this point, the material experiences structural changes, initiating deformation or irreversible alterations in its form.41) This parameter serves as a crucial indicator of a material’s mechanical limits and its ability to withstand external forces before exhibiting deformation or failure. This divergence can be attributed to the effect of crosslinking, which effectively prevents Ca-CAP from undergoing ionization. Consequently, Ca-CAP manifests superior mechanical strength compared to CAP. Furthermore, a noteworthy observation is the difference in the linear viscoelastic region (LVE) between the two polymers. CAP experiences a relatively shorter LVE, indicating a more limited range where its linear viscoelastic properties hold. In contrast, Ca-CAP exhibits a broader and more stable LVE. It’s also worth mentioning that CAP’s LVE shows irregular patterns and increased instability, further highlighting the impact of crosslinking on the rheological behavior of these polymers.
Frequency SweepIn the Frequency sweep analysis conducted on CAP and Ca-CAP polymers, their G′ values consistently surpassed G″ (G′ > G″), highlighting a prevalence of elastic behavior over viscosity. Within the Ca-CAP analysis, the frequency sweep plots distinctly showcased a substantial gap between G′ and G″ without any crossover, indicating the presence of a robust viscoelastic gel structure. This absence of crossover signifies enhanced stability for the Ca-CAP matrices owing to their distinct structural integrity.
In contrast, during the evaluation of the CAP matrix, a narrower margin between G′ and G″ was observed without any intersection, signifying a smaller difference between these parameters. This narrower gap implies a less pronounced viscoelastic structure and consequently denotes lower stability for the CAP matrix compared to Ca-CAP.42) This structural strength is attributed to the extensive entanglement in Ca-CAP matrices as a result of cross-linking. In contrast, CAP matrices were found to possess weaker gel structures when compared to their Ca-CAP counterparts. This phenomenon is attributed to the ionization of functional groups (–OCH2COO− H+) present in CAP, which induces electrostatic repulsion, consequently leading to a reduction in entanglement within the matrix.43,44)
According to Fig. 7a, the R2 value of 0.994 for CAP suggests a highly linear relationship between G′ and G,” indicating that the change and differences between the storage (G′) and loss (G″) moduli were minimal.45) This strong linear correlation implies that CAP exhibited less disparity between its elastic and viscous properties across the tested angular frequencies, resulting in a more balanced and less viscoelastic behavior.
On the other hand, the lower R2 value for Ca-CAP (0.848) indicates a less perfect linear relationship between G′ and G″ (Fig. 7b), signifying a larger disparity between its storage and loss moduli. This suggests that Ca-CAP showcased more significant differences between its elastic and viscous properties at various angular frequencies, demonstrating higher viscoelasticity.
The high R2 value for CAP implies a more uniform and balanced response between its elastic and viscous properties, resulting in a less viscoelastic behavior. Conversely, the lower R2 value for Ca-CAP indicates a more substantial difference between its storage and loss moduli, representing a higher degree of viscoelasticity with more pronounced differences between its elastic and viscous characteristics.
Furthermore, the best-fit slope values represent the rate of change of G″ with respect to G′. A higher slope value indicates a more pronounced viscous behavior relative to the elastic behavior. In this comparison, CAP demonstrated a substantially higher best-fit slope value of 0.662 ± 0.013 compared to Ca-CAP’s value of 0.254 ± 0.028. This indicates that CAP exhibits a steeper increase in G″ relative to G′, implying a more dominant viscous behavior compared to Ca-CAP.45)
The slope of the G″ vs. G′ plot was typically calculated as the change in G″ divided by the change in G′. It characterizes the relationship between the G′ and the G″ with respect to frequency, providing information about the viscoelastic behavior of the material. On the other hand, the loss tangent or phase angle (tanδ) was calculated separately as the ratio of G″ to G′. It represents the phase difference between the G′ and G″ responses of the polymers and provides insights into its viscoelastic properties.46)
Therefore, while both the slope value and the loss tangent are related to the viscoelastic behavior of the material, they are distinct measures and are calculated differently. The slope value characterizes the linear relationship between G″ and G′, while the loss tangent represents the phase difference between them.
In conclusion, based on the frequency sweep data analysis, it can be inferred that CAP exhibits stronger viscoelastic properties with a more prominent viscous behavior compared to Ca-CAP. This suggests that CAP is less elastic and more viscous than Ca-CAP under the conditions tested.
According to Table 4, the phase angle, represented as δ or the loss tangent (tanδ), crucially outlines a material’s viscoelasticity by comparing its loss and storage moduli (G″ and G′). This ratio unveils the balance between the elastic and viscous elements in a substance.40,47) Specifically, certain values of tanδ hold specific implications: when tanδ = 0, the material is ideally elastic; when tanδ = 100, the material is ideally viscous; when tanδ > 1, the material is more viscous than elastic; when tanδ < 1, the material is more elastic than viscous; and when tanδ = 1, the material is considered viscoelastic.48)
In particular, a higher phase angle suggests that the loss modulus (G″) dominates over the storage modulus (G′). This implies that the material dissipates more energy (associated with G″) than it stores (associated with G′). Therefore, a higher phase angle indicates a material that exhibits more pronounced viscous behavior, where energy is dissipated and stored energy is less readily recovered. This behavior contrasts with materials with lower phase angles, which tend to exhibit more elastic behavior, where energy is stored and recovered more efficiently.
In this dataset, studying CAP and Ca-CAP matrices phase angles across diverse angular frequencies reveals intriguing patterns. Both show lower phase angles at higher frequencies, indicating a stronger elastic response. As frequency drops, both matrices exhibit an upward trend in phase angle, hinting at a shift toward increased viscous behavior.47)
At 100 Rad/s, CAP sits around 0.63 in phase angle, while Ca-CAP is at 0.20. These lower values imply a robust elastic response in both polymers at higher frequencies. However, as frequency decreases, the phase angle ascends for both, showing a rising influence of the viscous component. At near-zero angular frequencies, CAP reaches roughly 0.30, whereas Ca-CAP is around 0.15. These higher phase angles at lower frequencies indicate a predominant viscous nature in both materials.40,47) These trends illustrate the viscoelastic changes in CAP and Ca-CAP concerning angular frequency. Their behavior leans elastic at higher frequencies and more viscous at lower frequencies. This shift in phase angles highlights the intricate interplay between their elastic and viscous properties, providing insights into their behavior across varying conditions.
Swelling and Erosion StudyIn the acidic medium, the degree of swelling observed in blank CAP tablets was notably lower than that observed in the buffer medium. Nevertheless, it is important to note that CAP tablets exhibited a rapid and more pronounced swelling in both acidic and buffer media when compared to Ca-CAP tablets. This disparity in swelling behavior is attributed to the calcium crosslinking process, which results in a reduced rate of water penetration (as indicated in Table 5) and a higher degree of entanglement within the Ca-CAP matrices. Consequently, this leads to decreased swelling in comparison to CAP matrices.20) In addition, Fig. 9 displays images of the CAP and Ca-CAP tablets retrieved after undergoing a 10-h (600-min) dissolution period.
Additionally, it was observed that the water penetration rate exhibited the following sequence: water penetration in a phosphate buffer (PB) solution > water penetration in an acidic solution. Furthermore, considering the water penetration velocity trend: PB solution > acidic solution, it follows that the rate of swelling also mirrored this sequence: swelling in PB solution > swelling in acidic solution. This higher degree of swelling and increased water penetration rate facilitated the dissolution of drugs within the matrix and their subsequent diffusion out.20) As a result, it can be anticipated that the release of the drugs would adhere to the order: release in PB solution > release in acidic solution. This is primarily due to the fact that the velocity of water penetration and the consequent swelling of a hydrophilic matrix depend on the characteristics of the gel layer that forms around the matrix tablet upon contact with water.
In the acidic medium, the extent of erosion observed in blank CAP tablets was significantly lower than that observed in the buffer medium. However, it is important to highlight that CAP tablets exhibited a higher degree of swelling in both acidic and buffer media when compared to Ca-CAP tablets. The presence of Ca2+ ions plays a crucial role in constraining the erosion of Ca-CAP matrix tablets.49) This phenomenon contributes to the sustained release characteristics of Ca-CAP matrices.
In-Vitro Drug Release StudyUpon the evaluation of the entire drug release profiles, it was observed that prolonged and increasingly sustained release characteristics were exhibited by Ca-CAP formulations with higher polymer concentrations. Conversely, a rapid momentary drug release profile was demonstrated by the CAP formulations. Notably, the maximum amount of DIL released from the CAP matrices was 120 mg (100%) within a 9-hour duration, while 119.964 mg (99.97%) of DIL was released from Ca-CAP matrices within 12 h (F7). This delay in drug release in Ca-CAP matrices was attributed to the restriction of water influx, reduced swelling, and the promotion of a viscoelastic gel layer formation due to Ca+2 crosslinking, contributing to a slower drug release. In CAP matrices, enhanced mobility, a disentangled structure, and the loss of structural integrity in weak gel networks facilitated rapid deformation due to electrostatic repulsion between the polymeric chains.50) Consequently, they exhibited increased water uptake, resulting in higher swelling and accelerated drug release.
The ANOVA table, as displayed in Table 8, confirmed that the differences in drug releases between the two matrices were statistically significant (p < 0.0001).51) The two-way ANOVA table examines the influence of two factors on the observed variation. In this analysis, the Row Factor contributes significantly, explaining about 26.93% of the total variation with a very low p-value (p = 0.0002), denoted by three asterisks (***), indicating its statistical significance. On the other hand, the Column Factor demonstrates a much smaller impact, accounting for 3.841% of the total variation, and possesses a non-significant p-value (p = 0.738), labeled as 'ns' (not significant). Overall, the Row Factor significantly affects the observed variation, whereas the Column Factor does not demonstrate a substantial impact, as indicated by the ANOVA results.
The results suggest that in vitro release profiles of all the matrix formulations of DIL-CAP conform better to the Korsmeyer Peppas model as the profiles showed better linearity when compared with other release kinetic equations. Release of drug from the matrix tablet generally follows the diffusion mechanism for water-soluble drugs. Fickian diffusion refers to the solute transport process in which the polymer relaxation time is much greater than the characteristic solvent diffusion time. In the case of the DIL-CAP tablet, values of exponent 'n' are found as 0.224–0.283 which indicates Case I Fickian diffusion. After burst release, the drug was released by diffusion through the gel layer formed around the tablet and initially huge burst release happened as polymer chains are not entangled much as the DIL-Ca-CAP tablet and diffusional resistance is less for the DIL-CAP tablet. Drug release characteristics of DIL-Ca-CAP tablets are somewhat different from that of DIL-CAP tablets. These profiles showed conformity with the Higuchi model and Korsmeyer–Peppas model (Table 9) as suggested by the regression coefficients. As water penetrates through the tablet a gel layer is formed on the surface of the tablet which acts like a thin film, and the drug is diffused from this thin layer. The exponent n values are closer to 0.45 except the n (0.381) value of F6. In the case of n > 0.45, the drug is released involving more than one mechanism (swelling/erosion) in addition to diffusion then it is categorized as anomalous diffusion, and when n ≤ 0.45, it is termed as Fickian diffusion.52) The formulation with a lesser amount of polymer (Ca-CAP) and a higher amount of disintegrant might have caused Fickian-type drug diffusion. There is the possibility of higher erosion when the amount of polymer is increased. Drug release rate is retarded successfully when ionically crosslinked carboxymethyl gum was used as the cumulative release is in the range of 52–60% during 6 h experiment.
Topography of Tablets by SEM StudiesIn Fig. 11B, the surface of the CAP tablet sample after 6 h of dissolution clearly shows significant erosion. Erosion here refers to the gradual breakdown of the tablet’s surface due to the dissolution process. Conversely, Fig. 11.D presents a contrasting image. The Ca-CAP tablet sample, at the same 0 to 6-h dissolution point, exhibits a notably different surface morphology (Figs. 11C, D). In this case, there is considerably less erosion, and instead, a gel-like structure has formed on the tablet’s surface. This gel formation indicates that the Ca-CAP tablet is undergoing a different dissolution behavior compared to the CAP tablet, resulting in a more controlled and sustained release of the drug. These SEM images provide visual evidence of the distinct dissolution characteristics between the two tablet formulations, further corroborating the findings related to drug release profiles and mechanisms.53,54)
Accelerated Stability Study of TabletsNotably, f2 values exceeding 50 signified that the dissolution profiles of the fresh and aged tablets remained highly consistent, indicating the stability of the drug and the formulations.55,56) Moreover, the stability analysis underscored that the characteristics of the Ca-CAP tablets remained unchanged throughout the storage period.
Conclusion
The comprehensive exploration into CAP and Ca-CAP polymers in drug delivery formulations has yielded valuable insights. FT-IR spectra revealed structural similarities with minor changes due to calcium ions. Enhanced O–H stretching in Ca-CAP suggests calcium crosslinking. DSC showed Ca-CAP’s thermal stability without any effect on the drug. Zeta potential analysis highlighted surface charge differences, and X-ray diffractograms confirmed higher crystallinity in Ca-CAP. Rheological studies unveiled distinct viscosities and mechanical behaviors attributed to cross-linking. Ca-CAP exhibited sustained drug release, while CAP showed rapid release following Fickian diffusion. SEM images illustrated the effect of dissolution on the surface texture of tablets. Stability studies affirmed the durability of drug-loaded tablets. In conclusion, calcium ion crosslinking significantly influences and culminates the properties and performance of Ca-CAP in sustained drug release behavior and heightened formulation stability, offering promising prospects for pharmaceutical applications.
Acknowledgments
We would also like to extend our gratitude to the Head of the Department of Pharmaceutical Technology at Jadavpur University.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1) Muruganantham S., Krishnaswami V., AnithaManikandan D, Aravindaraj N, Suresh J, Murugesan M, Kandasamy R. Gums as Pharmaceutical Excipients: An Overview. “Gums, Resins and Latexes of Plant Origin: Chemistry, Biological Activities and Uses,” Springer, Cham, 2022, pp. 145–189.
- 2) Pachuau L., Lalhlenmawia H., Mazumder B., Ind. Crops Prod., 40, 90–95 (2012).
- 3) Mukherjee S., Biswanath S. A., Khanam J., Karmakar S., Bhowmik R., International Journal of Drug Delivery Technology., 13, 551–561 (2023).
- 4) Jain A. K., Kumar N., Jain A., Garg R., Jain A., J. Drug Deliv. Ther., 8, 246–255 (2018).
- 5) Patel D. K., Purohit B. M., Joshi J. R., Natural polysaccharides in drug delivery and biomedical applications, ed. by Nasnain M. S., Nayak A. K., “Natural Polymers for Drug Delivery,” Academic Press, MA, 2019. pp. 1–33.
- 6) Kumar R., Rathi M., Yadav R., Sharma D., J. Drug Deliv. Sci. Technol., 54, 101348 (2019).
- 7) Yin X., Li H., Guo Z., Wu L., Chen F., de Matas M., Shao Q., Xiao T., York P., He Y., Zhang J., AAPS J., 15, 1025–1034 (2013).
- 8) Singh S., Srivastava M., Singh R., J. Pharm. Bioallied Sci., 13, 393–404 (2021).
- 9) Sharma R., Panda S. K., Devi S. M., Choudhury P. K., Int. J. Pharm. Investig., 8, 135–142 (2018).
- 10) Rajasekaran A., Sekar R., Duraipandiyan V., Ignacimuthu S., J. Young Pharm., 11, 342–346 (2019).
- 11) Mishra S. K., Patra S., Sahoo S. K., Saudi Pharm. J., 25, 398–405 (2017).
- 12) Huang Y., Yu H., Xiao C., Carbohydr. Polym., 66, 500–513 (2006).
- 13) Al-Hadeethi Y., Umar A., Ibrahim A. A., Al-Heniti S. H., Kumar R., Baskoutas S., Raffah B. M., Ceram. Int., 43, 6765–6770 (2017).
- 14) Phuong P. T., Oliver S., He J., Wong E. H., Mathers R. T., Boyer C., Biomacromolecules, 21, 5241–5255 (2020).
- 15) Fischer D., Li Y., Ahlemeyer B., Krieglstein J., Kissel T., Biomaterials, 24, 1121–1131 (2003).
- 16) Zhang X. Z., Li W. H., Gong X. L., Smart Mater. Struct., 17, 035027 (2008).
- 17) Shaayegan V., “Linear viscoelastic behaviour and relaxation phenomena of immiscible blends: the application of creep,” Doctoral dissertation, Concordia University, 2010.
- 18) Chaturvedi H., Garg A., Rathore U. S., Eur. J. Pharm. Med. Res., 4, 526–530 (2017), Internet.
- 19) Singh R., Maity S., Sa B., Carbohydr. Polym., 106, 414–421 (2014).
- 20) Ford J. L., Rubinstein M. H., McCaul F., Hogan J. E., Edgar P. J., Int. J. Pharm., 40, 223–234 (1987).
- 21) “Pharmacopoeia I,” Vol. II, The Controller Publication, Govt. of India, New Delhi, 2010, p. 1199.
- 22) Gubbi S. R., Jarag R., Asian J. Pharm. Sci., 5, 50–60 (2010).
- 23) Rao P. R., Diwan P. V., Drug Dev. Ind. Pharm., 24, 327–336 (1998).
- 24) Higuchi T. J., J. Pharm. Sci., 52, 1145–1149 (1963).
- 25) Korsmeyer R. W., Gurny R., Doelker E., Buri P., Peppas N. A., Int. J. Pharm., 15, 25–35 (1983).
- 26) Huang H., Wu Z., Qi X., Zhang H., Chen Q., Xing J., Chen H., Rui Y., Int. J. Pharm., 446, 211–218 (2013).
- 27) Coluccia S., Marchese L., Lavagnino S., Anpo M., Spectrochim. Acta A, 43, 1573–1576 (1987).
- 28) Dagar V., Pahwa R., Ahuja M., Biointerface Res. Appl. Chem., 13, 111 (2022).
- 29) Lu R., Gan W., Wu B. H., Zhang Z., Guo Y., Wang H. F., J. Phys. Chem. B, 109, 14118–14129 (2005).
- 30) Aguir C., M’Henni M. F., J. Appl. Polym. Sci., 99, 1808–1816 (2006).
- 31) Sumalatha B., Reddy D. V., Reddy D. S., Pharma Sci. Monitor, 6 (2015).
- 32) Krishan B. V., Rao C. H., Kishore V. S., Research Journal of Pharmacy and Technology., 13, 2315–2320 (2020).
- 33) Hongbo T., Yanping L., Min S., Xiguang W., Polym. J., 44, 211–216 (2012).
- 34) Vijayasankar GR, Bhargava A. Asian Journal of Pharmaceutical Science & Technology, 6, 19–26 (2016).
- 35) Liu Y., Lu K., Hu X., Jin Z., Miao M., Carbohydr. Polym., 234, 115908 (2020).
- 36) Bhatia M., Ahuja M., Int. J. Biol. Macromol., 72, 495–501 (2015).
- 37) Maiti S., Ray S., Mandal B., Sarkar S., Sa B., J. Microencapsul., 24, 743–756 (2007).
- 38) Huang Y., Yu H., Xiao C., Carbohydr. Polym., 66, 500–513 (2006).
- 39) Kaboorani A., Blanchet P., BioResources, 9, 4392–4409 (2014).
- 40) Ishikawa S., Kobayashi M., Samejima M., Chem. Pharm. Bull., 36, 2118–2127 (1988).
- 41) Chakravorty A., Barman G., Mukherjee S., Sa B., Carbohydr. Polym., 144, 50–58 (2016).
- 42) Talens P., Castells M. L., Verdú S., Barat J. M., Grau R., J. Food Eng., 292, 110265 (2021).
- 43) Medina-Torres L., Brito-De La Fuente E., Torrestiana-Sanchez B., Katthain R., Food Hydrocoll., 14, 417–424 (2000).
- 44) Yan X., Xiao X., Au C., Mathur S., Huang L., Wang Y., Zhang Z., Zhu Z., Kipper M. J., Tang J., Chen J., J. Mater. Chem. A Mater. Energy Sustain., 9, 21659–21684 (2021).
- 45) Lee J. H., Um C. M., Lee I. B., Dent. Mater., 22, 515–526 (2006).
- 46) Gao T., Gillispie G. J., Copus J. S., Pr A. K., Seol Y. J., Atala A., Yoo J. J., Lee S. J., Biofabrication, 10, 034106 (2018).
- 47) Ramli H., Zainal N. F., Hess M., Chan C. H., Chem. Teach. Int., 4, 307–326 (2022).
- 48) Wang H., Ke L., Ding Y., Rao P., Xu T., Han H., Zhou J., Ding W., Shang X., Food Hydrocoll., 122, 107079 (2022).
- 49) Wei X., Sun N., Wu B., Yin C., Wu W., Int. J. Pharm., 318, 132–138 (2006).
- 50) Nadgorny M., Ameli A., ACS Appl. Mater. Interfaces, 10, 17489–17507 (2018).
- 51) Nayak A. K., Pal D., Int. J. Biol. Macromol., 49, 784–793 (2011).
- 52) Korsmeyer R. W., Peppas N. A., J. Membr. Sci., 9, 211–227 (1981).
- 53) Murphy C., Pillay V., Choonara Y. E., du Toit L. C., Ndesendo V. M., Chirwa N., Kumar P., AAPS PharmSciTech, 13, 1–5 (2012).
- 54) Abe T., Yanagihara Y., Uchino T., Oriyama T., Komatsu M., Nakajima K., Suzuki H., Chem. Pharm. Bull., 62, 617–626 (2014).
- 55) Alshetaili A., Almutairy B. K., Alshehri S. M., Repka M. A., Pharmaceutics, 13, 213 (2021).
- 56) Aggarwal A. K., Jain S., Chem. Pharm. Bull., 59, 629–638 (2011).












