Abstract
Despite the great strides in biopharmaceuticals and monoclonal antibodies today, natural products remain highly attractive as drug candidates. Therefore, building a library of natural products through total synthesis is critically important for drug discovery. This perspective article details the collective total synthesis of polycyclic natural products using “bioinspired reactions” that mimic natural product biosynthesis. It discusses the total syntheses of 20 natural products, including dimeric diketopiperazine alkaloids, monoterpenoid indole alkaloids, and iridoid glycosides, each achieved in fewer than 14 steps starting from commercially available materials.
1. Introduction
Bioinspired transformations are chemical reactions in biosynthesis that are reproduced in flasks. In total synthesis, the term “biomimetic transformation or biomimetic reaction” has long been used.1,2) However, it is now only applied when the exact biosynthetic pathway is cleared by genetic engineering and mimicked. If a natural product chemist conceives a biosynthesis and reproduces the biosynthetic hypothesis in a flask, the terms “bioinspired” or “biogenetically inspired” would be appropriate. In 1917, a bioinspired total synthesis of tropinone as a synthetic precursor to atropine was achieved by Sir Robinson.3) This first bioinspired strategy, developed almost 100 years ago, demonstrated that natural products could be synthesized in a highly efficient and sophisticated manner. Since then, a succession of complex and fascinating natural products has been isolated, their biosynthetic hypotheses proposed, and several excellent bioinspired total syntheses have been achieved.2) Considering that approximately 50% of the medicines in use today are related to natural products, the appeal of bioinspired total synthesis has been and will continue to be unwavering.4) This review article describes the bioinspired total synthesis of polycyclic natural products developed by our group over the last decade. It particularly focuses on collective total synthesis, which, by leveraging biosynthetic hypotheses, can produce a large number of natural products, making it an efficient approach. This review highlights 20 natural product total syntheses achieved to date.
2. Bioinspired Total Syntheses of Tryptophan-Based Dimeric Diketopiperazine Alkaloids
A number of naturally occurring tryptophan-based dimeric diketopiperazine alkaloids have been isolated from Aspergillus, Streptomyces, and Eurotium species, which belong to the actinomycetes5) (Fig. 1). These molecules are unusual tetrapeptides consisting of four amino acids, including two tryptophan. It dimerizes at the C3 position of tryptophan, forming a pyrrolidinoindoline skeleton by nucleophilic cyclization of the Nb nitrogen to the C2 position of indole. Stereoisomers exist in nature at the C3 position. For example, WIN 64821 (1) and ditryptophenaline (2) are each diastereomer-like molecules, with a phenylalanine or N-methyl phenylalanine condensed to the tryptophan unit. In addition, several alkaloids have been isolated in which different amino acids are asymmetrically condensed onto each tryptophan unit (e.g., WIN 64745 (3)). Condensed amino acids are not only l-form but also D-form (e.g., asperdimin (4)). In addition, while the most commonly isolated dimeric molecules are C3 homodimers, heterodimers bound to the C3 position and other sites on the indole ring have also been isolated (e.g., naseseazine A (5)). At the time we began our synthesis, no clear biosynthetic pathway had been identified. Therefore, we started developing a bioinspired dimerization reaction, based on our biosynthesis hypothesis, to achieve a collective total synthesis of dimeric diketopiperazine alkaloids.
We assumed that biosynthesis occurs in water and, therefore, planned a dimerization reaction using radical processes that maintain high reactivity in aqueous conditions. Consequently, we investigated dimerization through radical coupling, aiming specifically at the generation of cation radicals in water via a one-electron oxidizing agent reacting on electron-rich indoles (Chart 1a). Methanesulfonic acid was added to dissolve the tryptophan ethyl ester, chosen as the substrate, in water. The reaction was carried out with various one-electron oxidizing agents. From our analysis, it was found that the desired dimerization via radicals proceeded when vanadium pentoxide and manganese acetate were used. When vanadium pentoxide was used, symmetrical isomers 7 and 8, produced by homocoupling at the C3 position, were obtained at yields of 28% each. By contrast, when manganese acetate was used as the oxidant, a heterodimer 9 with a C3-7 bond—similar to the bonding mode in the naseseazine series—was obtained as the major product. Both reaction conditions are effective on a decagram scale and do not require stringent conditions. Additionally, the reactions are conducted in water, with no organic solvents used. The key dimerized products, 7, 8, and 9, obtained on a gram scale, were respectively employed for the synthesis of diketopiperazine alkaloids. In this manuscript, the one-pot conversion processes of homodimer 7 to WIN 64821 (1) and WIN 64745 (3) are described (Chart 1b). WIN 64821 (1) is a diketopiperazine alkaloid formed by condensation followed by diketopiperazine ring formation of compound 7 with two l-phenylalanines. For the first condensation reaction, we have chosen to use 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM), developed by Ku-nishima et al.,6) which can form peptide bonds in alcohol or water. Thus, two equivalents of N-tert-butoxycarbonyl (Boc) phenylalanine and compound 7 were dissolved in ethanol, and DMT-MM was added to the condensation reaction. After the reaction, the solvent was removed under reduced pressure, and the crude materials were heated directly at 230°C (neat conditions). During this process, the Boc group was removed, and the diketopiperazine formation reaction proceeded. Finally, WIN 64821 (1) was obtained with a 70% yield in a one-pot process.7) The total synthesis of 1 was completed in just two one-pot reactions starting from the commercially available material and using only water and ethanol as solvents. Although diastereoselectivity was not observed in the initial dimerization reaction, the overall yield of the bioinspired total synthesis was 20%, making it a concise and efficient protocol. WIN64745 (5) is an alkaloid with an asymmetric structure, where l-leucine and l-phenylalanine are each condensed and cyclized to form a diketopiperazine ring. A stepwise condensation of each amino acid derivative took place next, followed by cyclization in one-pot, which required a close examination of the conditions. Thus, compound 7 was treated with 1 equivalent (equiv.) of N-benzyloxycarbonyl (Cbz) phenylalanine and ethyl 2-cyano-2-((dimethyliminio)-(morpholino)methyloxyimino)acetate hexafluorophosphate (COMU) as a condensing agent at low temperature. After 15 h of stirring, 1 equiv. of N-Cbz leucine was added to the reaction vessel. After an additional 12 h of stirring, the solvent was removed, methanol and a catalytic amount of palladium hydroxide were added, and the Cbz groups were removed under a hydrogen atmosphere. The reaction was then made basic with morpholine and stirred for 3 d. The intramolecular cyclization reaction proceeded, yielding the desired natural product 3 in a one-pot yield of 70% (total yield: 20%). Using similar condensation and cyclization reactions, a concise total synthesis of 11 tryptophan-based dimeric diketopiperazine alkaloids was achieved. After biological evaluation, some of these alkaloids were identified as new drug candidates for cancer therapy through USP7 inhibition and for atherosclerosis therapy through inhibition of macrophage formation.8,9)

Chart 1. Bioinspired Dimerization Reaction and Total Syntheses of WIN 64821 and WIN64745
3. Bioinspired Total Syntheses of Monoterpenoid Indole Alkaloids
Monoterpenoid indole alkaloids (MTIAs) are well-known representatives of plant alkaloids, with over 3000 species identified to date. Some of these, such as vinblastine and ajmaline, are already used as therapeutic drugs.10–15) In addition, numerous biological activities have been reported in the literature for these alkaloids in in vitro studies, highlighting their great potential for further development into pharmaceuticals. In biosynthesis, the condensation of tryptamine, derived from tryptophan, with secologanin (10)—a representative of secoiridoids—is followed by complex chemical transformations, leading to alkaloids with a variety of skeletons16–19) (Fig. 2). This class of alkaloids is widely recognized as monoterpenoid indole alkaloids. Due to their appealing structures and biological activities, these compounds have been the target of numerous total syntheses. It is easy to imagine that a large number of alkaloids could be prepared by mimicking this biosynthesis, starting from secologanin (10) and tryptamine. However, no total synthesis of secologanin (10) had been reported until the authors achieved it in 2019.20,21) Here, our concise total synthesis of secologanin (10) and the subsequent total syntheses of MTIAs (11–15) using bioinspired transformations will be described.
To achieve our planned collective total synthesis of MTIAs through bioinspired reactions, we must first develop a practical total synthesis of secologanin (10). The main challenges in synthesizing this compound lie in constructing the three contiguous chiral centers in the dihydropyran ring. Of the 3 chiral centers, the consecutive chiral carbons at C5 and C9 were constructed using our originally developed asymmetric organocatalytic Michael reaction (Chart 2). The electrophile 18 used in the key reaction was prepared in high yield via a Knoevenagel condensation of the commercially available propargylic aldehyde 16 with a thioester derivative 17 of malonic acid. The nucleophile used was sulfide-containing aldehyde 20, and the catalyst was diphenylprolinol silyl ether 19, known as the Hayashi–Jørgensen catalyst. The catalyst loading in this reaction was 3 mol%, with ether identified as the optimal solvent. The stereoselective Michael reaction proceeded through a transition state in which the alkyne side-chain adopted a gauche orientation relative to the catalyst moiety, resulting in the formation of adduct 21 with high conversion efficiency. To prevent isomerization at the α-position of the aldehyde moiety, 21 was subjected to Fukuyama reduction conditions22) without purification. During this reduction, the reaction proceeded selectively through the thioester, even in the presence of sulfides, which can poison Pd, and alkynes, which are sensitive to reduction reactions. The resulting aldehyde intermediate was immediately enolized to form a dihydropyran ring. The enantioselectivity of product 22 was measured at this stage and found to be over 99% enantiomeric excess. The chemical yield was 76% over 2 steps, making this a practical method that can be reliably performed on the decagram scale. The glycosylation reaction, which constructed 2 stereocenters, was accomplished using the Schmidt glycosylation method. Thus, hemiacetal compound 22 was treated with glycosyl trichloroacetimidate 23 at low temperature in the presence of BF3·Et2O to provide 24 in superb yield as a single isomer. This perfect stereoselectivity can be attributed to the steric hindrance from the 2 side chains on the dihydropyran ring and the neighboring effect of the acetyl group at the C2 position on the sugar chain. The synthesis of secologanin tetraacetate (25) was achieved through the removal of the TMS group, the construction of the aldehyde via hydroboration oxidation, and the formation of the terminal double bond by sulfoxide elimination. This compound 25 represents the key intermediate in our collective total synthesis using bioinspired transformations and can be prepared in 7 steps from commercially available products, with an overall yield of 25% on the decagram scale. Finally, acetyl groups on the sugar chain were removed via methanolysis, completing the first total synthesis of secologanin (10).20)
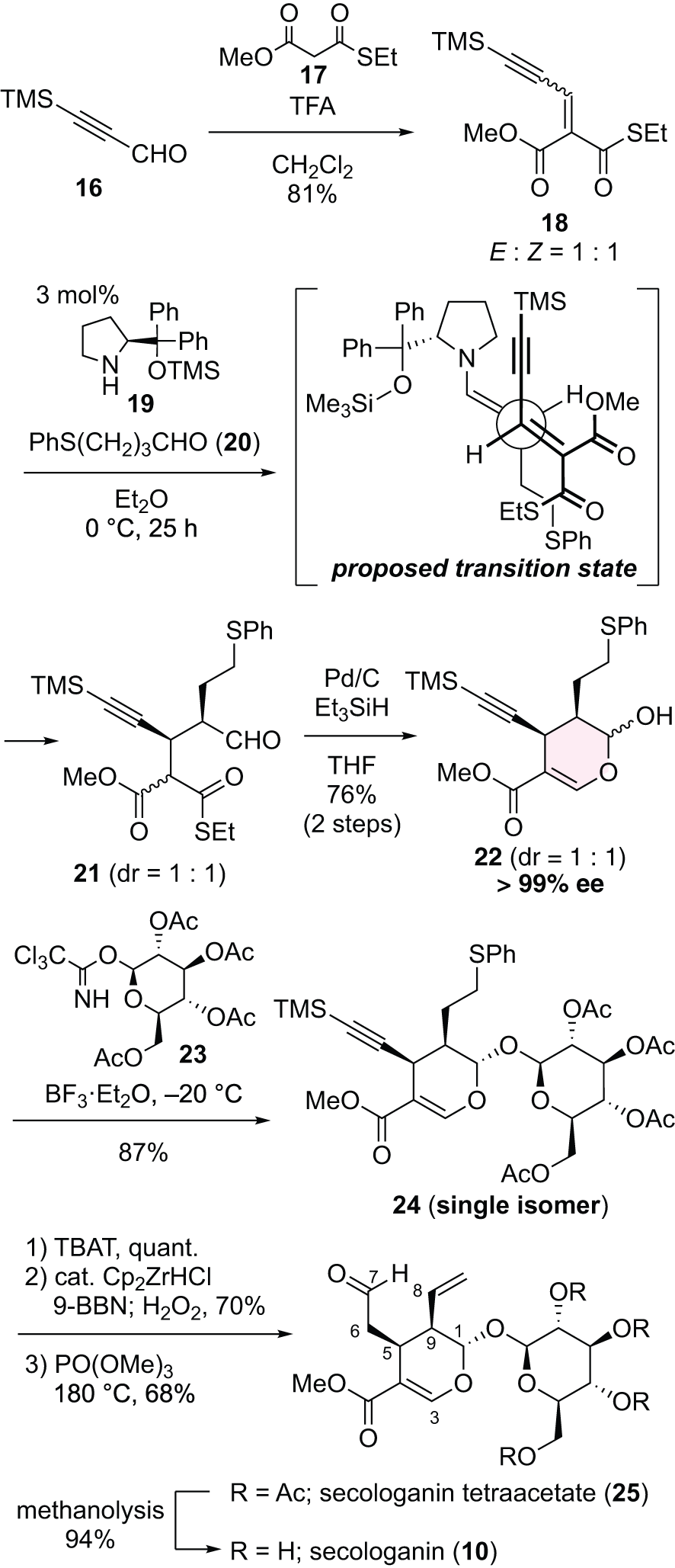
Chart 2. Total Synthesis of Secologanin
Strictosidine (11), obtained through the condensation reaction of secologanin (25) with tryptamine, is the first natural product in the biosynthesis of MTIAs. In the biosynthetic pathway, this intermediate is further converted into over 3000 alkaloids. Similar to secologanin (10), no one had completed the total synthesis of this important natural product at the time we began our synthetic study. We had already obtained secologanin tetraacetate (10), suggesting that strictosidine (11) could be easily synthesized through the Pictet–Spengler reaction with tryptamine. However, it was essential to control the newly generated C3 chiral center. Previous reports indicated that the enzymatic reaction using strictosidine synthase produced the C3S product.23) We attempted to reproduce this enzymatic reaction without the enzyme in a flask (Chart 3). Ultimately, we developed a new 2-step protocol using α-cyanotryptamine 26, derived from D-tryptophan, as a substrate. Thus, when secologanin derivatives 25 and 26 were stirred in the presence of trifluoroacetic acid (TFA), the reaction proceeded in just 3 min, yielding the adduct 27 quantitatively with a completely controlled conformation at the C3 chiral center. This remarkable stereoselectivity was demonstrated both experimentally and computationally. The acid-activated iminium ion forms a hydrogen bond with the carbonyl group at C22, leading to the formation of an 8-membered ring. During this process, the stereochemistry at C15 and the orientation of the cyano group at C5 cause the indole to approach from the upper side, thereby controlling the stereochemistry at C3. The cyano group at C5 can be reductively removed under acidic conditions, yielding strictosidine tetraacetate (28) in 85% yield. This diastereoselective Pictet–Spengler cyclization/decyanation sequence serves as an important alternative to strictosidine synthase. The first total synthesis of strictosidine (11) was achieved by removing the 4 acetyl groups of compound 28.24)

Chart 3. Total Synthesis of Strictosidine
After completing the total synthesis of strictosidine (11), we moved on to the total synthesis of cymoside (12), a glycosidic MTIA with a hexacyclic skeleton, isolated from Chimarrhis cymosa (Rubiaceae)25) (Chart 4). Even with the advancements of modern organic chemistry, constructing a cage ring structure that includes a propellane motif along with a continuous acetal arrangement and 8 contiguous chiral centers (excluding the sugar chain) cannot be easily achieved by conventional approaches. On the other hand, considering biosynthesis from strictosidine (11)—a potential precursor—makes this total synthesis achievable. In fact, the first total synthesis of cymoside (12) was achieved by Vincent and colleagues, and the bioinspired reaction was shown to be feasible.26) In our total synthesis, strictosidine derivative 27 was pretreated with TFA to suppress the nucleophilicity of the secondary amine by converting it into a salt. m-chloroperbenzoic acid was then added at 0°C, where it likely formed a hydrogen bonding network with the protons on the secondary amine, enabling stereoselective epoxidation of the C2–7 double bond of the indole core. The resulting epoxide was converted into an indolenine with an imine structure, yielding compound 29 with a β-hydroxy group introduced at C7. Intermediate 29 can be considered a key precursor in constructing the unique scaffold of cymoside (12). In fact, compound 29 was immediately transformed; a stereoselective oxa-Michael reaction occurred between the C7 hydroxyl group and C17, followed by a Mannich reaction involving the generated anion at C16 attacking the C2 position. This sequence constructed two 5-membered rings, resulting in the formation of the complex ring scaffold of natural product 12. The yield of the cascade reactions leading to compound 30 was 42%. Finally, under acidic conditions, the cyano group was reductively removed, and all acetyl groups on the sugar chain were removed by methanolysis, completing the total synthesis of cymoside (12). The overall yield of the synthesis was 7% over 11 steps, and this efficient approach was accomplished by mimicking the biosynthetic pathway.24)

Chart 4. Bioinspired Total Synthesis of Cymoside
In MTIAs, there are alkaloid glycosides with a β-carboline structure in which the C ring is oxidized. Most of these compounds are found in Rubiaceae plants, and some exhibit interesting biological activities, such as antileishmanial properties.27,28) Thus, we have performed the collective total synthesis of β-carboline alkaloids, which are more highly oxidized than strictosidine (11). The first synthetic target of β-carboline-type MTIAs is lyaloside (33), in which the C ring of strictosidine (11) is oxidized (Chart 5). In biosynthesis, it is assumed that the oxidation reaction proceeds from strictosidine itself, but replicating this reaction in a flask proved challenging. Therefore, we decided to utilize the cyano group of 27. When treated with silver nitrate as a Lewis acid, the cyano group was predominantly removed, and the resulting unstable enamine intermediate 31 was rapidly converted to the aromatic β-carboline 32 by air oxidation (94%). The resulting compound 32 was subjected to methanolysis, where all acetyl groups were removed, leading to the first total synthesis of lyaloside (33).29) We then turned our attention to the biosynthetically intriguing pentacyclic indole alkaloids, ophiorines A (13) and B (34), which feature unique intramolecular counterionic structures. For the direct conversion of lyaloside (33) to compounds 13 and 34, various additives and solvents were evaluated. As a result, heating compound 33 in an aqueous ammonium acetate solution for 3 d induced the aza-Michael reaction and subsequent hydrolysis of the methoxycarbonyl group, yielding the ionic natural products ophiorines A (13) and B (34) in 47 and 28% yields, respectively. Additionally, we have successfully achieved the total syntheses of two lyaroside-related β-carboline alkaloids.29)

Chart 5. Bioinspired Total Synthesis of Ophiorines A and B
MTIAs are known to achieve structural diversity through chemical transformations initiated by the cleavage of sugar chains during biosynthesis. In fact, most of the over 3000 identified MTIAs are alkaloids without sugar chains. We aimed to replicate these skeletal transformations in the early stage of MTIAs’ biosynthesis in the flask. Our initial approach was to synthesize secologanin aglycone silyl ether 36, in which the sugar chain is replaced by a silyl group (Chart 6). Silyl groups are more conveniently cleaved than sugar chains, as they can be removed under a variety of conditions, including acidic and fluoride anion conditions. The synthesis of secologanin aglycone 36 began with the silylation of the hydroxyl group on the hemiacetal of optically active cyclization product 22. When the bulky tert-butyldimethylsilyl (TBS) group was introduced in the presence of silver salts, the reaction proceeded selectively at the sterically accessible α-oriented hydroxyl group on the hemiacetal, similar to sugar chain introduction. Fortunately, the trimethylsilyl groups on the alkyne were also removed under these silylation conditions, yielding silyl ether 35 as a single isomer with 86% yield. Subsequent hydroboration/oxidation of the terminal alkyne to construct the aldehyde moiety, followed by the formation of the terminal olefin via sulfoxide elimination, completed the synthesis of secologanin aglycone silyl ether 36. As will be discussed later, hydrolysis of the methoxycarbonyl group of 36 was necessary for the bioinspired transformation. However, under standard hydrolysis conditions, this conversion was difficult due to the instability of both the aldehyde and the silyl group on the hemiacetal. This issue was ultimately resolved under aqueous triethylamine conditions, where it would typically not be employed in hydrolysis. Under these conditions, the aldehyde of 36 underwent hydration via intermediate 37 followed by intramolecular lactonization, yielding bicyclic compound 38 as a diastereomer mixture in excellent yield. The compound 38 could be used as a carboxylic acid equivalent under equilibrium conditions.
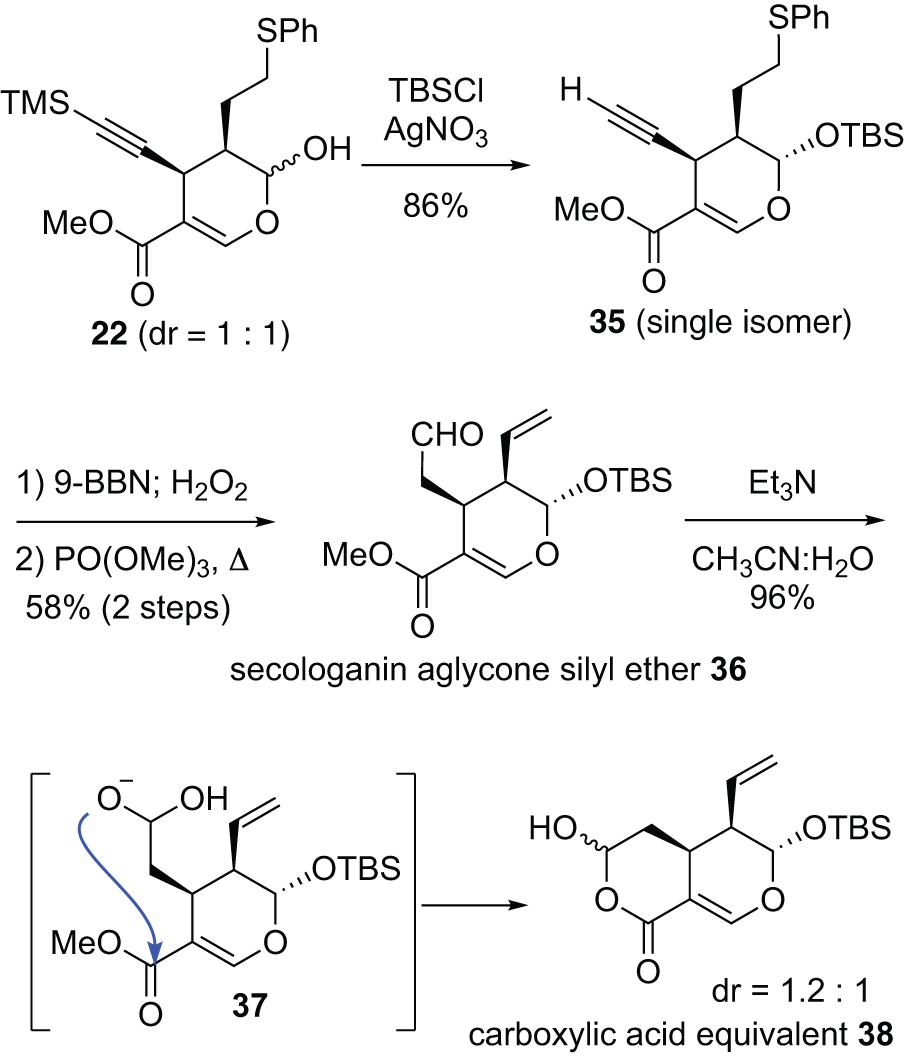
Chart 6. Synthesis of Secologanin Aglycone Silyl Ether
Considering that secologanin aglycones were available in sufficient quantities, their conversion to natural products through bioinspired reactions was investigated. First, the originally developed diastereoselective Pictet–Spengler reaction, followed by reductive decyanation, was carried out using α-cyanotryptamine (26) and secologanin aglycone silyl ether 36 to prepare the strictosidine aglycone silyl ether 39 (Chart 7a). The 2-step sequence proceeded with an 89% yield, and compound 39 was obtained as a single isomer. Then, compound 39 was treated with hydrochloric acid to remove the TBS group. As a result, the oxygen–silicon bond was not cleaved, but the siloxy group was eliminated and converted into an oxonium ion intermediate 40. Under acidic conditions, the secondary amine of 40 remains in its salt form, losing its nucleophilic properties. From the indole nitrogen of intermediate 40, the ring-closing reaction proceeded to the C21 position, resulting in the formation of nacycline (41) in 87% yield, which possesses a unique complex ring structure. Next, we attempted to remove the TBS group using fluoride anion. Thus, compound 39 was treated with tetrabutylammonium fluoride (TBAF) in acetic acid at –20°C. As expected, the silyl group was removed, and the resulting hemiacetal underwent ring-opening to form aldehyde intermediate 42. The secondary amine at N4 and the aldehyde at C21 of intermediate 42 cyclized to form dienamine intermediate 43. Equilibrium led to the formation of α,β-unsaturated iminium ion intermediate 44, and the oxa-Michael reaction proceeded from the nearby 17-position hydroxyl group to the C19 position, yielding the pentacyclic heteroyohimbine-type alkaloid cathenamine (45) in 67% yield. It can be concluded that the stereochemistry of C19 is controlled by the geometric isomerism of the C19–20 double bond, with the preferential formation of the thermodynamically stable Z-isomer being a result of this reaction. Because the natural product cathenamine (45) had an enamine structure and was unstable, sodium triacetoxyborohydride was added to the reaction vessel to perform the stereoselective reduction of the enamine in a one-pot reaction. As a result, tetrahydroalstonine (14) was obtained from strictosidine aglycone 39 in 77% yield. The total synthesis of 14, a representative heteroyohimbine-type alkaloid, was achieved in 10 steps with an overall yield of 21%.30)

Chart 7. Bioinspired Total Syntheses of Nacycline, Cathenamine, Tetrahydroalstonine, Akagerine, and Dihydrocycloakagerine
The bioinspired total synthesis of the akagerine-type indole alkaloids, found in Strychnos species, was subsequently performed (Chart 7b). The diastereoselective Pictet–Spengler reaction with α-cyanotryptamine (26) used the carboxylic acid equivalent 38 as the substrate. Subsequent decyanation yielded strictosidinic acid aglycone silyl ether 46. Further reductive methylation of the secondary amine at N4 produced carboxylic acid 47, the substrate for the bioinspired reaction, in 3 steps with an 87% yield. The silyl groups were then removed by TBAF. The resulting hemiacetal underwent ring-opening, and the resulting intermediate 48 was automatically decarboxylated. The C21 aldehyde was also converted into a thermodynamically stable α,β-unsaturated aldehyde through double bond isomerization. The resulting bisaldehyde 49 was then transformed into akagerine (50) by nucleophilic ring closure from the indole nitrogen to the C17 position (the nitrogen at N4 is masked by a methyl group). The yield of this impressive cascade reaction was 88%, although it is still unclear why the C17 aldehyde reacted selectively rather than the C21 aldehyde. Further reduction of the aldehyde at C21 of natural product 50 to allyl alcohol 51, followed by treatment with hydrochloric acid, resulted in ring closure from the C21 hydroxyl group to C17, affording the pentacyclic alkaloid dihydrocycloakgerine (52) in excellent yield. The total synthesis of 52 was accomplished in 13 steps from commercially available materials, with an overall yield of 21%, demonstrating high efficiency.30)
Kopsiyunnanine B (15) has a unique folded and complex pentacyclic structure containing 6 contiguous chiral centers.31) We proposed a biosynthetic pathway for this alkaloid and achieved its bioinspired total synthesis of 15 in the flask (Chart 8). Our proposed precursor for the bioinspired sequential cyclization reaction is the secondary amine compound 55. Thus, after the reduction of the aldehyde in secologanin aglycone 36 to primary alcohol 53, followed by a Mitsunobu reaction with tryptamine derivative 54 and removal of the nosyl group, compound 55 was prepared with high conversion. The key bioinspired reaction was achieved by initially treating with TBAF at low temperature, followed by acidification with acetic acid and gradually increasing the temperature to room temperature. The TBS group is first removed, yielding hemiacetal 56. Equilibrium then opens the hemiacetal to produce an electrophilic aldehyde at C21 (intermediate 57). Ring closure between the secondary amine at N4 and the aldehyde forms an iminium ion, while the double bond at positions 19–20 is converted to an electronically and thermodynamically stable α,β-unsaturated iminium ion, generating intermediate 59. A diastereoselective oxa-Michael reaction from the nearby C17 hydroxyl group proceeds, as in the case of heteroyohimbine type alkaloids, to yield the bicyclic compound 60. Finally, a diastereoselective Pictet–Spengler cyclization occurs, resulting in the pentacyclic compound 61 as the sole isolable product, with a yield of 45%. In this bioinspired cascade reaction, 3 new rings and 3 new chiral centers are constructed in a single step. The reduction of the β-acrylate moiety was achieved using l-selectride, with the stereochemistry at C16 controlled by kinetic protonation. Finally, after chlorination at the C7 position of compound 62 with NCS, an acid-mediated rearrangement in an aqueous solution was employed to construct the oxindole skeleton, completing the first total synthesis of kopsiyunnanine B (15).32)

Chart 8 Bioinspired Total Synthesis of Kopsiyunnanine B
This review article described the total synthesis of 11 MTIAs. After achieving efficient total syntheses of secologanin and its derivatives, several MTIAs were obtained through bioinspired reactions with remarkable efficiency.33–37) Notably, both total syntheses require fewer than 14 steps. Although not detailed in this section, we have completed the total synthesis of an additional 24 alkaloids (see, e.g., Fig. 3). For more details, please refer to the cited paper.
4. Bioinspired Total Syntheses of Hetero-Oligomeric Iridoid Glycosides
To date, more than 600 iridoid glycosides have been identified, primarily in plants.38,39) These iridoid glycosides, biosynthesized from isoprene, consist of 10 carbons and incorporate a number of oxygen atoms. The Dipsacus plant, long used in traditional medicine, contains numerous oligomeric iridoid glycosides40) (Fig. 4). These hetero-oligomeric iridoid glycosides (HOIGs) are expected to be active components responsible for the efficacy of traditional folk medicine. Most of the HOIGs isolated to date contain secologanin (10) and loganin (63) as monomeric units. Loganin (63) is a representative iridoid glycoside and serves as the precursor to secologanin (10) in biosynthesis.41) We aimed to accomplish a collective total synthesis of HOIGs, including cantleyoside (64), dipsaperine (65), (3R, 5S)-5-carboxyvincosidic acid 22-loganin ester (66), and dipsanoside A (67), starting with the total synthesis of loganin (63) via the reverse biosynthetic pathway.
Secologanin (10) is biosynthesized through an oxidative ring-opening reaction using loganin (63) as a substrate. Having succeeded in the large-scale synthesis of 10, we attempted the total synthesis of 63 via a reductive ring-closing reaction, which is the reverse of the biosynthetic process (Chart 9). Thus, secologanin tetraacetate (25) was treated with samarium iodide in the presence of hexamethylphosphoric triamide (HMPA), leading to a one-electron reduction of the carbonyl group, followed by a ring-closing reaction with an olefin to form the bicyclic loganin tetraacetate (68). The reaction proceeded in high yield, and the desired diastereoisomer was predominantly obtained (dr = 3 : 1 at C7). Following methanolysis to cleave the acetyl groups on the sugar chain, the total synthesis of loganin (63) was completed in a total of nine steps with an overall yield of 18%. On the other hand, to construct the oligomeric structure of HOIGs, the methoxycarbonyl group of secologin is required to be converted to a carboxylic acid. Thus, the aldehyde of compound 25 was temporally protected as an acetal, followed by hydrolysis of the methoxycarbonyl and acetyl groups. The carboxylic acid 69 was then prepared by re-acetylation of the sugar chain.

Chart 9. Total Synthesis of Loganin and Secologanic Acid Derivative
With the monomer synthesis complete, we proceeded with the total synthesis of the dimeric iridoid glycoside, cantleyoside (64) (Chart 10a). The synthesized secondary alcohol 68 and carboxylic acid 69 were coupled via a dehydration condensation reaction, followed by acetal deprotection to form an aldehyde, yielding cantleyoside octaacetate (70) in 67% over 2 steps. After the removal of all acetyl groups of 70, the first total synthesis of cantleyoside (64) was completed. This synthesis was accomplished in 11 steps from the commercially available materials, progressing through intermediate 68, with an overall yield of 12%.42)

Chart 10. Bioinspired Total Synthesis of Cantleyoside, Dipsaperine, (3R, 5S)-5-Carboxyvincosidic Acid 22-Loganin Ester, and Dipsanoside A
Next, we proceeded with the total synthesis of dipsaperine (65) and (3R, 5S)-5-carboxyvincosidic acid 22-loganin ester (66), both of which involve the condensation of the amino acid tryptophan with cantleyoside (64) (Chart 10b). Thus, compound 70, the protected form of cantleyoside (64), was coupled with l-tryptophan methyl ester via a Pictet–Spengler reaction under the acidic condition. The reaction proceeded in high yield, producing a separable mixture of diastereomers at the C3-position (3S:3R = 2 : 1). Hydrolysis of the methoxycarbonyl group in each diastereomer, along with the removal of the eight acetyl groups, was performed to achieve the total synthesis of the natural products 65 and 66. On the other hand, when compound 70 was treated with one equivalent of tryptophan under neutral conditions, a self-aldol condensation reaction selectively proceeded to form the E-isomer. In this reaction, the electronically stabilized E-olefin is likely preferentially formed. After the removal of all acetyl groups, the first total synthesis of the heterotetramer dipsanoside A (67) was achieved. The synthesis was completed in 12 steps with an overall yield of 9%.42) These condensation reactions from compound 70 to natural products 65, 66, and 67 could be ascribed to a bioinspired reaction.
5. Conclusion
Even in recent years, when biopharmaceuticals and monoclonal antibodies have dominated drug development, natural products remain important drug candidates. Therefore, the quantitative supply of promising natural products through total synthesis, followed by biological evaluation, is of utmost importance. This perspective article presented 20 examples of total syntheses of polycyclic natural products using bioinspired reactions, all of which have been achieved by our group over the past decade. In fact, we have successfully achieved the total synthesis of more than 50 natural products, though we were unable to include them in this article. Achieving the total synthesis of numerous natural products in a short period is not typically easy, but it is possible by considering their biosynthesis. In nature, natural products are biosynthesized from other natural products, allowing a single synthetic route to supply a wide variety of natural products. Since biosynthesis involves numerous reactions that significantly alter the skeleton, it also enables the generation of structural diversity. We will continue to expand our natural product library by fully utilizing biosynthesis-oriented, bioinspired reactions.
Acknowledgments
I would like to thank co-workers on the projects described herein for their continuous efforts, whose names are acknowledged in the publications from our group cited in the reference section.
Conflict of Interest
The author declares no conflict of interest.
Notes
This review of the author’s work was written by the author upon receiving the 2024 Pharmaceutical Society of Japan Award for Divisional Scientific Promotion.
References
- 1) Cordell G. A., “Introduction to Alkaloids: A Biogenetic Approach,” Wiley, New York, 1981.
- 2) Zografos A. L., “From Biosynthesis to Total Synthesis; Strategies and Tactics for Natural Products,” Wiley, New Jersey, 2016.
- 3) Robinson R., J. Chem. Soc. Trans., 111, 762–768 (1917).
- 4) Newman D. J., Cragg G. M., J. Nat. Prod., 83, 770–803 (2020).
- 5) Anthoni U., Christophersen C., Nielsen P. H., “The Alkaloids: Chemical and Biological Perspectives,” Vol. 13, ed. by Pelletier S. W., Chapter 2, Pergamon Press, London, 1999, pp. 163–236.
- 6) Kunishima M., Kawachi C., Iwasaki F., Terao K., Tani S., Tetrahedron Lett., 40, 5327–5330 (1999).
- 7) Tadano S., Mukaeda Y., Ishikawa H., Angew. Chem. Int. Ed., 52, 7990–7994 (2013).
- 8) Tadano S., Sugimachi Y., Sumimoto M., Tsukamoto S., Ishikawa H., Chem. Eur. J., 22, 1277–1291 (2016).
- 9) Review see; Tadano S., Ishikawa H., Synlett, 25, 157–162 (2014).
- 10) Cordell G. A., “Introduction to Alkaloids: A Biogenetic Approach,” Wiley, New York, 1981.
- 11) Pelletier S. W., “The Alkaloids: Chemical and Biological Perspectives,” Vol. 1, ed. by Pelletier S. W., Wiley, New York, 1983.
- 12) Phillipson J. D., Zenk M. H., “Indoles and Biogenetically Related Alkaloids,” Academic Press, London, 1980.
- 13) Saxton J. E., “The Chemistry of Heterocyclic Compounds, Indoles Part 4, The Monoterpenoid Indole Alkaloids,” Wiley, New York, 1983.
- 14) Saxton J. E., “The Chemistry of Heterocyclic Compounds, Supplement to Volume 25, Part 4, The Monoterpenoid Indole Alkaloids,” Wiley, New York, 1994.
- 15) Saxton J. E., Nat. Prod. Rep., 14, 559–590 (1997).
- 16) Smith G. N., Chem. Commun., 1968, 912–913 (1968).
- 17) Brown R. T., Smith G. N., Stapleford K. S. J., Tetrahedron Lett., 9, 4349 (1968).
- 18) Patthy-Lukát Á., Károlyházy L., Szabó L. F., Podányi B., J. Nat. Prod., 60, 69–75 (1997).
- 19) Patthy-Lukáts Á., Kocsis Á., Szabó L. F., Podányi B., J. Nat. Prod., 62, 1492–1499 (1999).
- 20) Rakumitsu K., Sakamoto J., Ishikawa H., Chem. Eur. J., 25, 8966–9000 (2019).
- 21) In 2021, Garg and coworkers reported total synthesis of secologanin and strictosidine; Anthony S. M., Tona V., Zou Y., Morrill L. A., Billingsley J. M., Lim M., Tang Y., Houk K. N., Garg N. K., J. Am. Chem. Soc., 143, 7471–7479 (2021).
- 22) Fukuyama T., Lin S. C., Li L., J. Am. Chem. Soc., 112, 7050–7051 (1990).
- 23) Cai Y., Zhu H., Alperstein Z., Yu W., Cherkasov A., Zou H., ACS Chem. Biol., 12, 3086–3092 (2017).
- 24) Sakamoto J., Umeda Y., Rakumitsu K., Sumimoto M., Ishikawa H., Angew. Chem. Int. Ed., 59, 13414–13422 (2020).
- 25) Lémus C., Kritsanida M., Canet A., Genta-Jouve G., Michel S., Deguin B., Grougnet R., Tetrahedron Lett., 56, 5377–5380 (2015).
- 26) Dou Y., Kouklovsky C., Gandon V., Vincent G., Angew. Chem. Int. Ed., 59, 1527–1531 (2020).
- 27) Levesque J., Pousset J. L., Cave A. C. R., Seances Acad. Sci., Ser. C., 280, 593 (1975).
- 28) Aimi N., Murakami H., Tsuyuki T., Nishiyama T., Sakai S., Haginiwa J., Chem. Pharm. Bull., 34, 3064–3066 (1986).
- 29) Sakamoto J., Hiruma D., Kitajima M., Ishikawa H., Synlett, 34, 576–581 (2024).
- 30) Sakamoto J., Ishikawa H., Chem. Eur. J., 28, e2021104052 (2022).
- 31) Wu Y., Kitajima M., Kogure N., Zhang R., Takayama H., Tetrahedron Lett., 49, 5935–5938 (2008).
- 32) Imaoka S., Nakashima Y., Kitajima M., Ishikawa H., Chem. Pharm. Bull., 72, 68–74 (2024).
- 33) Nakashima N., Sakamoto J., Rakumitsu K., Kitajima M., Juliawaty L. D., Ishikawa H., Chem. Pharm. Bull., 70, 187–191 (2022).
- 34) Sakamoto J., Kitajima M., Ishikawa H., Chem. Pharm. Bull., 70, 662–668 (2022).
- 35) Sakamoto J., Kitajima M., Ishikawa H., Chem. Eur. J., 29, e202300179 (2023).
- 36) Review; Sakamoto J., Ishikawa H., Synlett, 34, 2293–2303 (2023).
- 37) Review; “New Tide of Natural Product Chemistry,” ed. by Ishikawa H., Takayama H., Chapter 10, Sakamoto J., Ishikawa H., pp. 211–234, Springer, Singapore, 2023.
- 38) Boros C. A., Stermitz E. R., J. Nat. Prod., 53, 1055–1147 (1990).
- 39) Boros C. A., Stermitz E. R., J. Nat. Prod., 54, 1173–1246 (1991).
- 40) Tao Y., Chen L., Yan J., J. Ethnopharmacol., 258, 112912 (2020).
- 41) Yamamoto H., Katano N., Ooi A., Inoue K., Phytochemistry, 53, 7–12 (2000).
- 42) Yoshidome A., Sakamoto J., Kohara M., Shiomi S., Hokaguchi M., Hitora Y., Kitajima M., Tsukamoto S., Ishikawa H., Org. Lett., 25, 347–352 (2023).



