Results and Discussion
Design and Synthesis of KNT-127 Derivatives
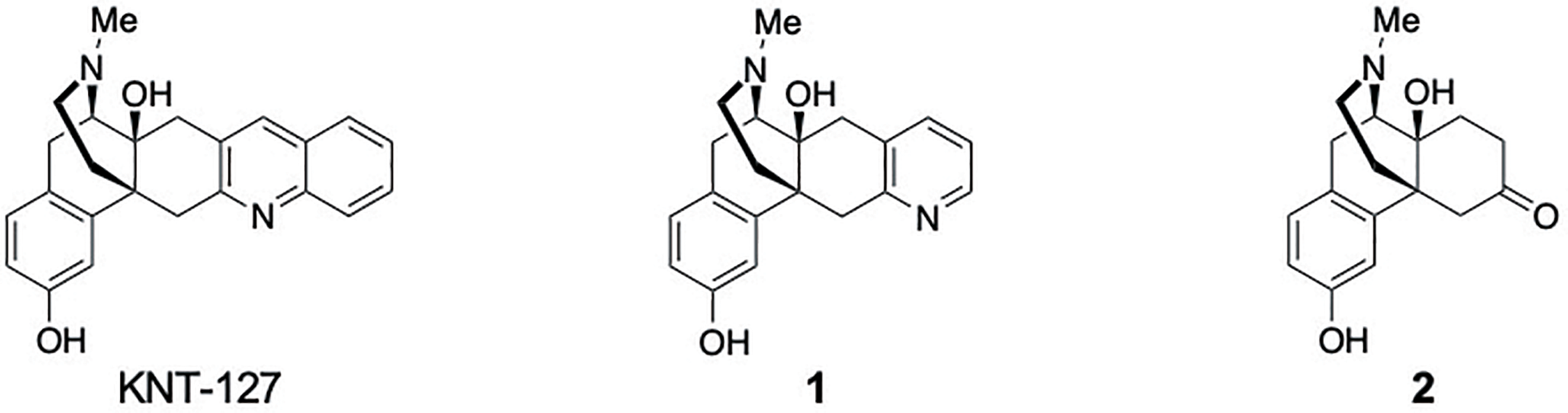
KNT-127 was originally designed based on the structure of (–)-TAN-67, which itself was derived from the DOR antagonist naltrindole (NTI)29) by removing the 10-methylene bridge and the 4,5-epoxy moiety and replacing the indole structure with quinoline as the key address component responsible for DOR selectivity.14) Despite the importance of the quinoline ring in DOR-selective agonist activity, its precise contribution toward receptor selectivity and signal bias between the G-protein and β-arrestin pathways has not been thoroughly investigated to date. To address this shortcoming, we designed novel morphinan derivatives including the pyridine-containing morphinan derivative 1 and the ketone-containing morphinan derivative 2 by systematically removing aromatic rings from the quinoline moiety of KNT-127 (Fig. 2).
As shown in Chart 1, the synthetic route to 1 and 2 began with commercially available naltrexone hydrochloride (3), which was converted into intermediate 10 through a 9-step literature procedure15,30) (Chart 1A). Etherification of 3 using methyl iodide (MeI) furnished methyl ether 4 in 92% yield. The 4,5-epoxy ring of 4 was cleaved with zinc powder to afford phenol 5 in 90% yield. A copper-catalyzed Ullmann coupling between 5 and bromobenzene provided 6, and its subsequent acetalization generated cyclic ketal 7 (85% yield over 2 steps). The phenyl ether group of 7 was removed via a Birch reduction, yielding 8 in 91% yield. After protecting the hydroxy group of 8 with an acetyl group, the cyclopropyl methyl group on the tertiary amine was removed and the amine group simultaneously protected using trichloroethoxycarbonyl-Cl (Troc) to obtain carbamate 9 (80% yield over 2 steps). LiAlH4 reduction of the carbamate in 9 and simultaneous deacetylation, followed by removal of the acetal group afforded intermediate 10 (64% yield over 2 steps). With intermediate 10 in hand, we then synthesized the KNT-127 derivatives (Chart 1B). The quinoline ring of 11 was constructed using a modified Friedländer synthesis, followed by demethylation to yield KNT-127 (88% yield over 2 steps). The geometric isomerism of the quinoline ring in KNT-127 was determined based on heteronuclear multiple bond correlations (HMBCs) using two-dimensional-NMR (2D-NMR) spectroscopy and induced accordingly at the C6 and C7 positions of the morphinan skeleton (Supplementary Fig. S1). For pyridine derivative 1, a copper-catalyzed cyclization31) of propargylamine and intermediate 10 was performed to construct the pyridine ring, yielding 12 in 35% yield. The structure of 12 was also confirmed based on HMBCs using 2D-NMR spectroscopy, which established the presence of a C6, 7-pyridine structure (Supplementary Fig. S2). Finally, pyridine derivative 1 was obtained in 53% yield by demethylation of 12. The ketone derivative 2, devoid of any aromatic rings, was synthesized in 55% yield by direct demethylation of intermediate 10.
Binding Assay
With the synthesized derivatives in hand, we first evaluated their binding affinity for the opioid receptors (MOR, DOR, and KOR) using a competitive binding assay with radiolabeled ligands (Table 1 and Supplementary Fig. S3). Compared to KNT-127 (Ki = 0.16 nM, MOR/DOR = 237, KOR/DOR = 483), pyridine derivative 1 exhibited a 15-fold decrease in DOR affinity (Ki = 2.36 nM), while its affinity for MOR and KOR was significantly increased, resulting in reduced receptor selectivity (MOR/DOR = 2.22, KOR/DOR = 18.3). Ketone derivative 2 showed a further 10-fold decrease in DOR affinity (Ki = 22.6 nM) compared to the pyridine derivative, and a 141-fold decrease compared to KNT-127, with a corresponding 23- and 5-fold increase in MOR and KOR affinity, respectively, leading to a complete loss of DOR selectivity (MOR/DOR = 0.07, KOR/DOR = 0.65). These results suggest that the quinoline ring of KNT-127 acts as a multifunctional site, enhancing DOR affinity while decreasing affinity for other opioid receptors, thus supporting its role as an address component in DOR-selective agonists.15)
Table 1. Binding Affinity of KNT-127 and Its Derivatives
1 and
2a)
 |
| Compounds |
Binding affinity, Ki (nM) (95% CI)a) |
Selectivity ratio |
| MORb) |
DORc) |
KORd) |
MOR/DOR |
KOR/DOR |
| Met-Enk |
2.52
(1.13–5.61) |
0.52
(0.34–0.81) |
373.0
(159–878) |
4.85 |
717 |
| KNT-127 |
37.9
(9.97–144) |
0.16
(0.12–0.22) |
77.2
(47.9–124) |
237 |
483 |
| 1 |
5.23
(1.56–17.5) |
2.36
(1.69–3.39) |
43.1
(27.5–67.5) |
2.22 |
18.3 |
| 2 |
1.67
(0.76–3.69) |
22.6
(12.4–41.0) |
14.7
(7.54–28.6) |
0.07 |
0.65 |
a) Ki values were calculated based on receptor-binding assay results using HEK293 cell membranes. Cell membranes were obtained from HEK293 cells stably expressing each human opioid receptor. Data are presented as the mean (95% confidence interval [95% CI]) calculated from 2 independent experiments, each performed in triplicate. b) [3H]-DAMGO was used. c) [3H]-DPDPE was used. d) [3H]-U-69593 was used.
To investigate the effect of the quinoline ring on signaling bias, specifically Gi-protein dissociation and βarr2 recruitment through the activation of the DORs, we used a split NanoLuc Binary Technology (NanoBiT) assay (Fig. 3 and Table 2).32–34) KNT-127, as a full agonist, demonstrated robust G-protein-dissociation activity (EC50G = 2.70 nM, Emax = 94.0%), but only partial agonist activity for βarr2-recruitment activity (EC50β = 15.9 nM, Emax = 37.3%). These results are consistent with previous reports highlighting the efficacy-dominant G-protein-biased DOR agonism of KNT-127.24) Conversion of the quinoline to a pyridine ring resulted in a 25.4-fold reduction (p < 0.0001, Supplementary Table S1) in potency for Gi-protein activation (1; EC50G = 63.2 nM, Emax = 71.1%), along with a 23% decrease in efficacy compared to 1 (p = 0.0018, Supplementary Table S2). In contrast, the βarr2-recruitment activity of 1 (EC50β = 121 nM, Emax = 9.40%) showed a 7.8-fold reduction in potency (p = 0.0007, Supplementary Table S3), and a 73% decrease in efficacy (p = 0.0027, Supplementary Table S4). Further removal of another aromatic ring (2; EC50G = 937 nM, Emax = 73.0%) led to a 376-fold decrease in Gi-protein activation potency (p < 0.0001, Supplementary Table S1) and a 21% decrease in efficacy (p = 0.0029, Supplementary Table S2). However, βarr2-recruitment activity was significantly diminished (2; EC50β = NA, Emax = NA). These results indicate that while the partial agonist activity for Gi-protein activation was largely retained despite the removal of the aromatic rings from 1, both potency and efficacy for βarr2 recruitment were significantly reduced. Based on these results, the bias factor (ΔΔLog(τ/KA))35,36) of each compound relative to KNT-127 was calculated. A comparison of the bias factors between KNT-127 and Met-Enk, as previously reported,24) confirmed that KNT-127 exhibits reduced preference for βarr2 recruitment and is a G-protein-biased agonist. The removal of 1 aromatic ring from KNT-127 resulted in a ΔΔLog(τ/KA) value close to zero (1; ΔΔLog(τ/KA) = –0.034), indicating a signaling preference similar to KNT-127. In contrast, the removal of 2 aromatic rings (2) eliminated detectable βarr2-recruitment activity, making bias factor evaluation impossible.
Table 2. NanoBiT-Based Gi-Protein-Dissociation and
βarr2-Recruitment Assays for Met-Enk, KNT-127, and Its Derivatives
1 and
2
| Compounds |
Gi-protein dissociation |
βarr2 recruitment |
Bias factor |
| EC50 (nM) |
Emax (%)a) |
EC50 (nM) |
Emax (%)b) |
ΔΔLog(τ/KA)c) |
| Met-Enk |
1.26 ± 0.20ns |
100 ± 1.21ns |
33.2 ± 1.83* |
100 ± 4.47*** |
–0.23 |
| KNT-127 |
2.70 ± 0.60 |
94.0 ± 1.95 |
15.9 ± 2.57 |
37.3 ± 2.37 |
0 |
| 1 |
63.2 ± 5.08*** |
71.1 ± 2.55** |
121 ± 16.6*** |
9.40 ± 0.64** |
–0.034 |
| 2 |
937 ± 80.2*** |
73.0 ± 5.80** |
NA |
NA |
— |
a) Gi-protein dissociation Emax was calculated as a relative value at the reference ligand (Met-Enk) as 100%. b) βarr2 recruitment Emax was calculated as a relative value with Span at the reference ligand (Met-Enk) as 100%. c) ΔΔLog(τ/KA) of Met-Enk and 1 were compared to KNT-127. Data are presented as mean ± S.E.M. from 3 independent experiments, each performed in duplicate. Statistical analysis was performed using one-way ANOVA followed by Dunnett’s test. ns, Not significant; *p < 0.05; **p < 0.01; ***p < 0.001 compared to KNT-127. NA, parameter not available owing to a lack of ligand response.
Furthermore, Gi-protein-dissociation activity of these derivatives was also evaluated for MOR and KOR. Consistent with the binding-assay results, both 1 (EC50G = 83.0 nM, Emax = 95.0%) and 2 (EC50G = 11.0 nM, Emax = 100%) exhibit more potent agonist activity for MOR than KNT-127 (EC50G = 240 nM, Emax = 96.0%), while showing minimal agonist activity for KOR. 2 exhibited MOR/DOR dual agonist activity, while 1 exhibited MOR-selective agonist activity (Fig. 4 and Table 3). These findings suggest that the extension of the aromatic rings toward the horizontal direction of the morphinan skeleton is critical not only for DOR-selective binding affinity but also for agonist activity.
Table 3. NanoBiT-Based Gi-Protein-Dissociation Assays for MOR, DOR, and KOR with Met-Enk, KNT-127, and Its Derivatives
1 and
2
| Compounds |
MOR |
Selectivity ratio
MOR/DORb) |
KOR |
Selectivity ratio
KOR/DORb) |
| EC50 (nM) |
Emax (%)a) |
EC50 (nM) |
Emax (%)b) |
| Met-Enk |
3.34 ± 0.33 |
100 ± 1.86 |
3.2 |
1900 ± 305 |
100 ± 5.23 |
1844 |
| KNT-127 |
246 ± 28.9 |
96.1 ± 1.67 |
182 |
2442 ± 53.2 |
46.4 ± 2.01 |
1808 |
| 1 |
84.4 ± 9.09 |
95.1 ± 1.89 |
1.8 |
554 ± 24.5 |
19.4 ± 1.58 |
23 |
| 2 |
11.3 ± 1.11 |
100 ± 0.34 |
0.02 |
247 ± 18.7 |
40.0 ± 1.32 |
0.40 |
a) Gi-protein dissociation Emax was calculated as a relative value at the reference ligand (Met-Enk) as 100%. b) The receptor selectivity ratios were calculated using the EC50 values for DOR obtained from Table 2. Data are presented as mean ± S.E.M. from 3 independent experiments, each performed in duplicate.
The results of this study provide valuable insights into the structure–signal relationship of the quinoline moiety in KNT-127, a DOR-selective agonist. The systematic removal of aromatic rings from the quinoline moiety significantly altered the pharmacological profile of the derivatives, both in terms of receptor binding and intracellular signaling pathways. The pyridine and ketone derivatives showed a drastic reduction in DOR affinity and selectivity, highlighting the critical role of the quinoline ring in maintaining DOR selectivity. This suggests that the quinoline moiety serves as an address domain, enhancing the binding interactions required for DOR selectivity. Recent advances in the structural analysis of opioid receptors complexed with endogenous opioids and synthetic opioids have elucidated that the extracellular vestibule, encompassing the extracellular loops (ECL2 and ECL3) and the extracellular termini of TM2, TM6, and TM7, is not conserved in terms of sequence and charge distribution among opioid-receptor subtypes.37,38) This region functions as a selectivity filter for various ligands and plays a crucial role in fine-tuning the efficacy of receptor activation by different ligands.
In the crystal structure of the DOR bound to the agonist DPI-287,26) the 4-N,N-diethylcarbamoylbenzene moiety of DPI-287 occupies a sub-pocket formed by the V6.55, W6.58, and L7.35 residues between TM6 and TM7. This sub-pocket is unique to the DOR, as it is composed of different residues in MOR (V6.55, K6.58, and W7.35) and KOR (I6.55, E6.58, and Y7.35). The bulkier aromatic residues in MOR and KOR likely induce steric hindrance with the N,N-diethyl carbamoyl moiety of DPI-287, and thus DPI-287 selectively binds to DOR. Similarly, the DOR antagonist NTI also occupies this sub-pocket with its indole moiety, and this specific interaction underpins its DOR selectivity.39) The terminal benzene ring of the quinoline moiety in KNT-127 may also occupy the same V6.55W6.58L7.35 sub-pocket in DOR. Our results show that the removal of the terminal benzene ring of KNT-127 resulted in a significant decrease in DOR affinity while enhancing the affinity for MOR and KOR. This suggests that the interaction with the V6.55W6.58L7.35 sub-pocket is crucial for DOR selectivity, that is, the absence of the terminal benzene ring of KNT-127 likely reduces steric interactions with the bulky residues in MOR and KOR, thereby increasing the affinity of the KNT derivative to these receptors. Thus, the interaction of the quinoline moiety with the V6.55W6.58L7.35 sub-pocket represents the essence of the address component of the DOR selectivity of KNT-127, a hypothesis that has been experimentally confirmed for the first time in this study.
In terms of signaling pathways, the systemic removal of the fused bicyclic ring in KNT-127 significantly affected both Gi-protein activation and βarr2 recruitment. While the potency for G-protein signaling decreased with the removal of the aromatic rings, the efficacy was largely preserved, indicating that the core morphinan structure is sufficient to maintain G-protein activation. On the other hand, the β-arrestin recruitment was profoundly affected, that is, both the potency and efficacy showed substantial reductions. This result indicates that the quinoline moiety, particularly its extended aromatic structure, would play a role in not only the enhancement of potency for G-protein signaling but also that of the efficacy of β-arrestin recruitment. These findings are consistent with previous reports that have shown that KNT-127 and (-)-TAN-67 exhibit partial agonist activity for β-arrestin recruitment, which has been suggested to be linked to their reduced side-effect profile.
The structural divergence between the mechanisms of G-protein activation and β-arrestin recruitment underscores the complexity of achieving selective modulation of intracellular pathways. The quinoline moiety, by interacting with specific extracellular residues, enhances both DOR selectivity and the partial agonism for β-arrestin recruitment, while the core morphinan structure is responsible for G-protein activation. This study, therefore, provides a structural framework for understanding how modifications to the morphinan skeleton can selectively modulate receptor-signaling pathways. Such insights would be crucial for the rational design of biased agonists that retain therapeutic efficacy while minimizing adverse effects.
Recent findings have questioned the role of β-arrestin signaling in the adverse effects of MOR agonists.36,40,41) While early studies in βarr2-knockout mice implicated β-arrestin in MOR-mediated tolerance and respiratory depression,42) newer evidence indicates that morphine and fentanyl still induce these side effects in βarr2-knockout mice, suggesting alternative mechanisms.43) In addition, partial MOR agonists with low efficacy for G-proteins also show reduced β-arrestin efficacy while increasing the therapeutic window for respiratory depression.44) Lutz et al. further demonstrated that MOR/DOR G-biased dual partial agonists with moderate but submaximal efficacy exhibit antinociceptive effects with only limited respiratory depression as an adverse effect.45) These findings highlight the complex interplay between G-protein and β-arrestin signaling in opioid pharmacology, suggesting that improvements in the safety profile may be attributable either to reduced β-arrestin-mediated signaling or to intrinsic low efficacy across all pathways. In this context, 2 exhibits partial DOR agonist activity, offering insights for designing DOR partial agonists. Zhang et al. have reported that N-methylmorphinan derivatives with indole or 2′-amino-thiazole substituents at the 6,7-position exhibit partial agonist activity against DOR in GTPγS binding assays.46) These findings, combined with the present study’s results, suggest that modulating both the direction and distance of the additional aromatic motif may serve as a novel design strategy to shift agonist activity toward partial agonism, not only for the G-protein pathway but also for β-arrestin signaling. These insights provide a valuable framework for developing safer, more effective DOR-targeting therapeutics.
This study has several limitations that should be acknowledged. First, the conclusions are based on two derivatives (1 and 2) of KNT-127. While these derivatives provide meaningful insights into the role of the morphinan skeleton and quinoline moiety, a broader range of analogs would allow for a more comprehensive understanding of the structure–signal relationships. Additional derivatives with modifications to the quinoline moiety will be reported in a subsequent study along with their synthesis and pharmacological characterization.
Second, the absence of in vivo behavioral pharmacological testing is a notable limitation. Such experiments are essential for validating the therapeutic potential and safety profiles of these compounds. Despite this limitation, the current study provides a robust in vitro framework for understanding the structural determinants of DOR selectivity and signaling bias. Future research will prioritize in vivo evaluations to further elucidate the pharmacological properties of these derivatives.
Finally, computational docking and molecular dynamics simulations were not included in this study. Docking analyses of KNT-127 and its derivatives have been conducted based on the modifications to the quinoline moiety and will be reported in a subsequent study. These simulations will further clarify the interactions between the quinoline moiety and the DOR, offering mechanistic insights that complement the experimental findings.
Despite these caveats, the findings of this study lay the groundwork for future research aimed at optimizing the therapeutic profiles of DOR-selective agonists through structural modifications. These insights advance our understanding of the molecular mechanisms underlying DOR selectivity and biased signaling.
Experimental
Chemistry
General Information
All chemical reagents and solvents were purchased from the following commercial suppliers: Tokyo Chemical Industry (Tokyo, Japan), Sigma-Aldrich, Inc. (St. Louis, MO, U.S.A.), Kanto Chemical Co., Inc. (Tokyo, Japan), Wako Pure Chemical Corporation, Ltd. (Osaka, Japan), and Nacalai Tesque (Kyoto, Japan). All commercially available chemicals and solvents were used without further purification. The synthetic compounds described in this study were checked using analytical TLC (Merck Co., Ltd., Darmstadt, Germany), Kieselgel 60 F254, 0.25 mm), visualized under UV light at 254 nm using phosphomolybdic acid in an aqueous solution of sulfuric acid, Hanessian stain, ninhydrin, or p-anisaldehyde followed by heating. Column chromatography was carried out on silica gel (a: spherical, neutral, 40–50 µm, Kanto Chemical Co.; b: spherical, neutral, CHROMATOREX PSQ60B, 60 µm, Fuji Silysia Chemical Ltd., Aichi, Japan). Preparative TLC was performed on Kieselgel 60 F254 plates (Merck Co., Ltd., 0.50 mm). Melting points (mp) were determined on a Yanaco MP mp apparatus and are uncorrected. Optical rotations were measured with an Anton Paar MCP 100 Polarimeter. Infrared spectra were recorded on a JASCO FT/IR 4100Plus spectrophotometer. 1H- and 13C-NMR spectral data were obtained using JEOL JNM-ECS 400 instruments. Chemical shifts are quoted in ppm using CDCl3 (δ 7.26 ppm), CD3OD (δ 3.31 ppm), C5D5N (δ 7.55 ppm), or tetramethylsilane (TMS; δ 0 ppm) as the reference for the 1H-NMR data. CDCl3 (δ 77.16 ppm), CD3OD (δ 49.00 ppm), and C5D5N (δ 135.50 ppm) were used as references for the 13C-NMR data. Signal patterns are indicated as br (broad peak), s (singlet), d (doublet), or m (multiplet). 1H-NMR chemical shifts were assigned using a combination of correlation spectroscopy (COSY), nuclear Overhauser effect spectroscopy (NOESY), and heteronuclear single quantum coherence (HSQC) data. Similarly, 13C-NMR chemical shifts were assigned based on heteronuclear multiple bond connectivity (HMBC) and HSQC experiments. Mass spectra were measured using a JEOL JMS-T100LP (ESI-TOF) mass spectrometer. The purity (≥95%) of the assayed compounds was determined via analytical HPLC or elemental analysis. Analytical HPLC was performed on an AQUITY ultra-performance liquid chromatography (UPLC) system (Waters Co., Ltd., Tokyo, Japan) equipped with an AQUITY UPLC BEH C18 column (1.7 µm, 50 mm × 2.1 mm); PDA detection at 210–400 nm; column temperature: 40°C. Elemental analyses were performed on a J-SCIENCE LAB Micro Corder JM10.
Synthesis of the Target Compounds and Their Spectroscopic Data
(4bR,8aS,9R)-8a-Hydroxy-3-methoxy-11-methyl-8,8a,9,10-tetrahydro-5H-9,4b-(epiminoethano)phenanthren-6(7H)-one (10)
A solution of 9 (5.0 g, 9.11 mmol) in tetrahydrofuran (THF) (40 mL) was added to a stirred suspension of LiAlH4 (3.5 g, 91.1 mmol) in THF (100 mL) at 0°C under an Ar atmosphere. Then, the mixture was warmed to room temperature. After 2 h of stirring, a saturated aqueous solution of Rochelle’s salt (200 mL) was added to the solution, before it was extracted with EtOAc (300, 200, and then 100 mL). The combined organic extracts were washed with brine, dried over Na2SO4, and filtered, before the filtrate was concentrated under reduced pressure. A solution of the resulting compound in 1 M HCl (100 mL) was stirred for 1 h at 60°C under an Ar atmosphere before it was basified (pH = 9) with a saturated aqueous solution of NaHCO3 (150 mL), and extracted with CHCl3 (300, 200, and then 100 mL). The combined organic extracts were dried over Na2SO4 and filtered, before the filtrate was concentrated under reduced pressure. The thus obtained residue was purified by column chromatography on silica gel (1–10%, 10% NH3 aq./MeOH in CHCl3) to afford 10 (1.76 g, 64%) as a pale orange amorphous solid.
1H-NMR (400 MHz, CDCl3) δ: 7.01 (d, J = 8.2 Hz, 1H), 6.80 (d, J = 2.8 Hz, 1H), 6.70 (dd, J = 8.2, 2.8 Hz, 1H), 4.73 (brs, 1H), 3.77 (s, 3H), 3.16 (d, J = 18.3 Hz, 1H), 3.04 (d, J = 14.2 Hz, 1H), 2.84–2.71 (m, 4H), 2.40 (s, 3H), 2.36 (m, 1H), 2.18–2.08 (m, 3H), 1.89–1.76 (m, 2H), 1.18 (m, 1H); 13C-NMR (100 MHz, CDCl3) δ: 210.3, 158.5, 140.5, 128.6, 127.2, 112.7, 111.1, 69.3, 62.3, 55.4, 46.5, 45.2, 45.1, 42.9, 37.7, 36.9, 31.9, 23.9. The spectral data are in agreement with those reported in the literature.49)
(6R,6aS,14aR)-17-Methyl-5,6,7,14-tetrahydro-6aH-6,14a-(epiminoethano)naphtho[2,1-b]acridine-2,6a-diol (KNT-127)
A mixture of 10 (300 mg, 0.951 mmol), 2-aminobenzaldehyde (230.1 mg, 1.90 mmol), and 2% KOH/EtOH (2.0 mL) was placed in a sealed microwave vial under an Ar atmosphere. The reaction mixture was heated to 110°C at a pressure of 2 bar under microwave irradiation for 50 min. After cooling to room temperature, the reaction mixture was poured into water (10 mL) and extracted with CHCl3 (40, 30, and then 20 mL). The combined organic extracts were dried over Na2SO4 and filtered, before the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (1–10%, 10% NH3 aq./MeOH in CHCl3) to afford 11 (336 mg, 92%) as a colorless amorphous solid.
A stirred solution of 11 (330 mg, 0.854 mmol) in CH2Cl2 (17 mL) at 0°C under an Ar atmosphere was treated with 1.0 M BBr3 in CH2Cl2 (3.42 mL, 3.42 mmol). After 30 min of stirring, the reaction mixture was basified (pH = 9) using an aqueous solution of ammonia (30%, 6.0 mL), and extracted with CHCl3 (60, 50, and then 40 mL). The combined organic extracts were dried over Na2SO4 and filtered, before the filtrate was concentrated under reduced pressure. The thus obtained residue was purified by column chromatography on silica gel (1–10%, 10% NH3 aq./MeOH in CHCl3) to afford KNT-127 (308 mg, 96%) as a colorless amorphous solid (purity >95%, determined by UPLC at UV max).
1H-NMR (400 MHz, CD3OD) δ: 7.89 (d, J = 8.2 Hz, 1H), 7.84 (s, 1H), 7.71 (m, 1H), 7.63 (ddd, J = 8.7, 6.9, 1.4 Hz, 1H), 7.44 (ddd, J = 8.2, 6.9, 0.9 Hz, 1H), 6.96 (d, J = 8.2 Hz, 1H), 6.74 (d, J = 2.3 Hz, 1H), 6.51 (dd, J = 8.2, 2.3 Hz, 1H), 3.64 (d, J = 17.4 Hz, 1H), 3.46 (d, J = 17.4 Hz, 1H), 3.28 (m, 1H), 3.14 (d, J = 17.9 Hz, 1H), 3.01 (d, J = 6.9 Hz, 1H), 2.98–2.90 (m, 2H), 2.51 (m, 1H), 2.46 (s, 3H), 2.32–2.23 (m, 2H), 1.37 (m, 1H), 2 protons (OH) were not observed; 13C-NMR (100 MHz, CD3OD) δ: 158.8, 157.0, 147.0, 141.9, 137.5, 130.2, 129.9, 128.8, 128.3, 128.0, 127.8, 127.0, 115.1, 112.8, 79.5, 70.9, 63.1, 46.7, 43.2, 41.5, 39.6, 37.6, 37.2, 25.0. The spectral data are in agreement with those reported in the literature.15)
(6R,6aS,12aR)-2-Ethoxy-15-methyl-5,6,7,12-tetrahydro-6aH-6,12a-(epiminoethano)naphtho[2,1-g]quinolin-6a-ol (12)
Propargyl amine (121 µL, 1.90 mmol) was added to a mixture of 10 (49.8 mg, 0.165 mmol) and CuCl2·2H2O (16.6 mg, 97.4 µmol) in EtOH (0.66 mL) at room temperature under ambient atmosphere. The mixture was heated to reflux for 24 h before it was concentrated under reduced pressure. The thus obtained residue was diluted with CHCl3 (10 mL), treated with EDA (0.50 mL) and H2O (10 mL), and vigorously stirred for 15 min at room temperature. Then, the mixture was extracted with CHCl3 (3 × 30 mL), and the combined organic layers were dried over Na2SO4 and filtered, before the filtrate was concentrated under reduced pressure. The thus obtained residue was purified by column chromatography on silica gel (1–5% MeOH in CHCl3) to afford 12 (19.7 mg, 35%) as a yellow amorphous solid.
[α]D20: –170.0 (c = 0.20, CHCl3); IR (NaCl): 2925, 1448, 1243, 1042 cm–1; 1H-NMR (400 MHz, CDCl3) δ: 8.30 (d, J = 4.1 Hz, 1H), 7.19 (d, J = 7.6 Hz, 1H), 7.00 (d, J = 8.2 Hz, 1H), 6.93 (dd, J = 7.6, 5.0 Hz, 1H), 6.84 (d, J = 2.3 Hz, 1H), 6.63 (dd, J = 8.2, 2.3 Hz, 1H), 3.66 (s, 3H), 3.49 (d, J = 17.4 Hz, 1H), 3.36 (d, J = 17.4 Hz, 1H), 3.26–3.19 (m, 1H), 2.92–2.86 (m, 3H), 2.68 (d, J = 17.4 Hz, 1H), 2.49–2.38 (m, 4H), 2.23–2.13 (m, 2H), 1.37 (m, 1H), 1 proton (OH) was not observed; 13C-NMR (100 MHz, CDCl3) δ: 158.4, 155.3, 146.9, 141.4, 136.7, 129.6, 128.5, 127.7, 121.2, 112.4, 110.6, 69.4, 61.8, 55.3, 45.7, 43.1, 40.5, 38.7, 36.7, 36.0, 24.1; HR-MS (ESI): m/z [M + H]+ (Calcd for C21H25N2O2+: 336.19160, found: 336.19197).
(6R,6aS,12aR)-15-Methyl-5,6,7,12-tetrahydro-6aH-6,12a-(epiminoethano)naphtho[2,1-g]quinoline-2,6a-diol (1)
A stirred solution of 12 (20.0 mg, 0.0594 mmol) in CH2Cl2 (1.2 mL) at 0°C under an Ar atmosphere was treated with 1.0 M BBr3 in CH2Cl2 (0.36 mL, 0.36 mmol). After 1.5 h of stirring, the reaction mixture was basified (pH = 9) with an aqueous solution of ammonia (30%, 1.0 mL), and extracted with CHCl3 (20, 10, and then 5 mL). The combined organic extracts were dried over Na2SO4 and filtered, before the filtrate was concentrated under reduced pressure. The thus obtained residue was purified using preparative TLC (10%, 10% NH3 aq./MeOH in CHCl3) to afford 1 (10.2 mg, 53%) as a colorless amorphous solid (purity >95%, determined by elemental analysis).
mp 306–308°C; [α]D20: –182.0 (c = 0.20, CHCl3/MeOH = 10:1); IR (KBr): 3376, 2942, 1446, 1243, 1055 cm–1; 1H-NMR (400 MHz, C5D5N) δ: 11.26 (brs, 1H), 8.45 (d, J = 4.6 Hz, 1H), 7.40 (d, J = 2.3 Hz, 1H), 7.14–7.11 (m, 2H), 6.98 (dd, J = 8.2, 2.3 Hz, 1H), 6.86 (dd, J = 7.8, 4.6 Hz, 1H), 3.78 (d, J = 16.9 Hz, 1H), 3.64 (d, J = 16.9 Hz, 1H), 3.22 (d, J = 17.9 Hz, 1H), 3.07 (d, J = 17.4 Hz, 1H), 2.95–2.85 (m, 2H), 2.83 (d, J = 17.4 Hz, 1H), 2.28–2.16 (m, 6H), 1.29 (m, 1H), 1 proton (OH) was not observed; 13C-NMR (100 MHz, C5D5N) δ: 157.4, 156.3, 146.9, 142.1, 136.5, 130.5, 129.2, 126.8, 121.4, 114.7, 112.8, 69.7, 62.0, 45.8, 42.9, 40.7, 39.5, 36.9, 36.6, 24.3; HR-MS (ESI): m/z [M + H]+ (Calcd for C20H23N2O2+: 323.17595, found: 323.17576); Anal. Calcd for C20H22N2O2: C, 70.94; H, 7.08, N, 8.27. Found: C, 71.04; H, 7.11, N, 8.10.
(4bR,8aS,9R)-3,8a-Dihydroxy-11-methyl-8,8a,9,10-tetrahydro-5H-9,4b-(epiminoethano)-phenanthren-6(7H)-one (2)
A stirred solution of 10 (997 mg, 3.31 mmol) in CH2Cl2 (33 mL) at –20°C under an Ar atmosphere was treated with 1.0 M BBr3 in CH2Cl2 (23 mL, 23 mmol). After 30 min of stirring, the reaction mixture was basified (pH = 9) with an aqueous solution of ammonia (30%, 15 mL), and extracted with CHCl3 (50, 30, and then 20 mL). The combined organic extracts were dried over Na2SO4 and filtered, before the filtrate was concentrated under reduced pressure. The thus obtained residue was purified using column chromatography on silica gel (1–10%, 10% NH3 aq./MeOH in CHCl3) to afford 2 (526 mg, 55%) as a colorless amorphous solid (purity >95%, determined by UPLC at UV max).
1H-NMR (400 MHz, CDCl3) δ: 6.96 (d, J = 8.2 Hz, 1H), 6.83 (d, J = 2.8 Hz, 1H), 6.65 (d, J = 8.2, 2.8 Hz, 1H), 3.14 (d, J = 18.3 Hz, 1H), 3.06 (d, J = 14.2 Hz, 1H), 2.84–2.69 (m, 4H), 2.39–2.36 (m, 4H), 2.18–2.09 (m, 3H), 1.91–1.77 (m, 2H), 1.18 (d, J = = 9.6 Hz, 1H), 2 protons (OH) were not observed; 13C-NMR (100 MHz, CDCl3) δ: 212.0, 154.9, 140.4, 128.9, 126.8, 114.3, 112.6, 69.3, 62.3, 46.5, 45.14, 45.10, 42.9, 37.7, 36.9, 31.9, 23.9. The spectral data are in agreement with those reported in the literature.49)
Bioassays
Competitive Binding Assay
The tested compound was dissolved in dimethyl sulfoxide (DMSO) to prepare a 10 mM stock solution (10 µL), which was then diluted with 1990 µL of assay buffer (50 mM Tris, 1 mM ethylenediaminetetraacetic acid (EDTA), 5 mM MgCl2) to achieve a 50 µM working solution. Moreover, 200 µL of the working solution was serially diluted with 1800 µL of assay buffer containing 0.5% DMSO to achieve final concentrations as low as 5 pM. Subsequently, a membrane suspension obtained from HEK293 cells stably expressing the opioid receptors (MOR, DOR, or KOR) was incubated in 250 µL of assay buffer with various concentrations of the tested compound and 2 nM tritiated opioid radioligand ([3H]-DAMGO for MOR, [3H]-DPDPE for DOR, and [3H]-U69593 for KOR: PerkinElmer, Inc., Waltham, MA, U.S.A.) at 25°C for 2 h with gentle shaking at 300 rpm. The reaction was terminated by filtration using a Filtermat B glass filter (PerkinElmer, Inc.) with a FilterMate cell harvester (PerkinElmer, Inc.). The filters were washed 3 times with 50 mM Tris buffer, and then dried for 70 min at 60°C. Finally, MeltiLex B/HS (PerkinElmer, Inc.) was melted on the dried filter for 5 min at 90°C. The radioactivity in the filter was determined with a Microbeta scintillation counter (PerkinElmer, Inc.). Nonspecific binding was measured in the presence of 10 µM unlabeled opioid ligand (DAMGO for the MORs, DPDPE for the DORs, and U-69593 for the KORs). Sigmoidal concentration-response curves and Ki values were calculated according to the Cheng–Prusoff equation using the Prism software package (Version 8.4.3; GraphPad Software Inc., La Jolla, CA, U.S.A.). Data are presented as the mean (95% confidence interval [95% CI]) calculated from 2 independent experiments, each performed in triplicate.
Intracellular Signaling Assays
Reagents and Plasmids
Human full-length DOR was N-terminally fused to the FLAG-epitope tag with the preceding hemagglutinin (HA)-derived signal sequence (MKTIIALSYIFCLVFADYKDDDDKGGSGGGGSGGSSSGGG; FLAG epitope tag underlined); the resulting construct is referred to as ssHA-FLAG-DOR. Plasmids for the NanoBiT-based G-protein-dissociation assay32) and the bystander NanoBiT-based β-arrestin-recruitment assay33,34) have been described previously. Unless otherwise indicated, all the constructs were inserted into the pCAGGS-expression-plasmid vector.
Cell Culture and Transfection
HEK293A cells (Thermo Fisher Scientific) were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Nissui Pharmaceutical) supplemented with 5% fetal bovine serum (Gibco, Thermo Fisher Scientific), 100 units/mL penicillin (Sigma-Aldrich), 100 µg/mL streptomycin (Gibco, Thermo Fisher Scientific), and 2 mM l-glutamine (Gibco, Thermo Fisher Scientific) (complete DMEM). Transfection was performed using polyethylenimine (PEI) solution (Polyethylenimine “Max,” Polysciences). Typically, HEK293A cells were seeded in a 10 cm culture dish at a cell density of 2 × 105 cells/mL in 10 mL of the complete DMEM and cultured for 1 d in a humidified incubator (37°C; 5% CO2). A transfection solution was prepared by combining a plasmid solution diluted in 500 µL of Opti-MEM and 500 µL of Opti-MEM solution containing 25 µg of PEI. The transfected cells were further incubated for 1 d before being subjected to assays as described below.
NanoBiT-Based G-Protein-Dissociation Assay
Ligand-induced G-protein dissociation was measured using the NanoBiT-based G-protein-dissociation assay,32) in which the interaction between a Gα-subunit and a Gβγ-subunit was monitored by the NanoBiT system (Promega). Specifically, the interaction between the Gαi1 subunit fused with a large fragment (LgBiT) at the α-helical domain (between residues 91 and 92 of Gαi1; Gαi1-LgBiT) and the N-terminally small fragment (SmBiT)-fused Gγ2 subunit with a C68S mutation (SmBiT-Gγ2-CS) was examined. HEK293 cells in a 10-cm dish were transfected with a mixture of 1000 ng of ssHF-DOR plasmid, 500 ng of Gα-LgBiT plasmid, 2500 ng of Gβ1 plasmid, and 2500 ng of SmBiT-Gγ2 (C68S) plasmid. After incubation for 24 h, the transfected cells were collected with 0.53 mM EDTA-containing phosphate-buffered saline, centrifuged at 190 × g for 5 min, and suspended in 2 mL of Hanks’ balanced salt solution containing 0.01% bovine serum albumin (BSA; fatty-acid-free grade; SERVA) and 5 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid) (pH = 7.4) (assay buffer). The cell suspension was dispensed in a white 96-well plate at a volume of 80 µL per well and loaded with 20 µL of 50 µM coelenterazine (Carbosynth or Angene) diluted in the assay buffer. After 2 h of incubation at room temperature, the plate was measured for baseline luminescence (SpectraMax L, Molecular Devices), and 20 µL of 6× ligand diluted in the assay buffer, or the assay buffer alone (vehicle) was manually added. The plate was read for 15 min with an integration time of 0.18 s per read and an interval of 20 s at room temperature. The luminescence counts over 10–15 min after ligand addition were averaged and normalized to the initial counts. Concentration–response curves were calculated by dividing the luminescence change at each concentration point by that of the vehicle-added condition.
Bystander NanoBiT-Based βarr2-Recruitment Assay
Ligand-induced β-arrestin recruitment to the plasma membrane was measured using the bystander NanoBiT-based β-arrestin-recruitment assay,33,34) in which the interaction between β-arrestin and a plasma-membrane-anchored split luciferase was monitored by the NanoBiT system (Promega). Specifically, the interaction between the N-terminally small-fragment (SmBiT)-fused βarr2 and the plasma-membrane-localized large fragment of the split-luciferase (LgBiT-CAAX) was examined. A plasmid encoding LgBiT-CAAX was constructed by fusing a human KRAS-derived CAAX motif (GKKKKKKSKTKCVIM) to the C-terminus of LgBiT via a flexible linker (GGSGGGGSGGSSSGG). HEK293 cells in a 10-cm dish were transfected with a mixture of 1000 ng of ssHF-DOR plasmid, 500 ng of SmBiT-β-arrestin and 2500 ng of LgBiT-CAAX plasmid. The transfected cells were subjected to the same procedure as described in the “NanoBiT-based G-protein-dissociation assay” section.
Statistical Analysis
Statistical analysis procedures were performed using the GraphPad Prism 10 software package (GraphPad). For the analysis of the pharmacological parameters, agonist-induced responses were fitted to a 4-parameter sigmoidal concentration–response curve with the Hill slope constrained to an absolute value less than 1.5 using the following equation: Y = Bottom + (Top – Bottom)/(1 + 10((LogEC50 – X) × Hill Slope))) (GraphPad Prism 10). Maximum effect (Span) values (“Top” – “Bottom”) and pEC50 values (negative logarithmic values of half-maximum effective concentration [EC50] values) were calculated from the obtained concentration–response curves. Emax was calculated as a relative value with the Span at the reference ligand set to 100%.
Bias factor (ΔΔLog(τ/KA)) was calculated as described previously.44) The τ and KA values were determined by fitting the concentration–response data to the operational model50) using GraphPad Prism 10 software (GraphPad).




