Abstract
Glucose-dependent insulinotropic polypeptide (GIP), secreted from enteroendocrine K cells, has potent insulin-releasing and extrapancreatic glucoregulatory activities. However, exogenous GIP has less potent biological effects compared with another incretin hormone, GLP-1, which limits its use for the treatment of type 2 diabetes. The fate and secretion of administered native GIP remain unclear. The aim of this study was to identify plasma binding proteins for human GIP. Fluorescent-labelled GIP was added to fresh human plasma and subjected to clear native polyacrylamide gel electrophoresis (CN-PAGE). Then fluorescent protein bands were in-gel trypsin-digested and subjected to liquid chromatography tandem-mass spectrometry (LC-MS/MS) analysis, revealing the presence of albumin, immunoglobulin G (IgG) and transferrin. In contrast to GIP, the binding of fluorescent GLP-1 and glucagon to plasma protein fractions were minimal. CN-PAGE analysis of synthetic GIP incubated with human serum albumin, purified IgG or transferrin, and subsequent western blot analysis revealed that GIP binds to each of these proteins. Taken together, these results indicate that GIP readily binds to albumin, IgG and transferrin, three plasma proteins highly abundant in the human peripheral circulation. Separation of protein complexes using CN-PAGE and the identification of in-gel digested proteins by LC-MS/MS analysis provide a promising strategy to identify plasma binding proteins for bioactive peptides.
GLUCOSE-DEPENDENT INSULINOTROPIC POLYPEPTIDE (GIP) is a 42-amino acid polypeptide secreted from duodenal and small intestinal K-cells [1]. GIP, an incretin hormone, inherently contributes to postprandial blood glucose homeostasis by potentiating the glucose-dependent release of insulin from pancreatic β cells [2, 3]. In addition, GIP has numerous important extrapancreatic effects associated with its receptor expression located in adipose, brain, bone, the cardiovascular system, and gastrointestinal tract [4]. Thus, GIP might have a critical role in some of the key pathophysiologic characteristics of type 2 diabetes. However, the actions of GIP administered to healthy subjects and type 2 diabetes patients are clearly diminished compared with GLP-1, which retains its potency as an insulinotropic factor in type 2 diabetes [5, 6]. A thorough review of the recent literature revealed that most data regarding GIP secretion is inconsistent and confusing despite the extensive study of the secretory mechanisms and endogenous degradation of GIP [7].
One important unappreciated question is whether GIP readily and abundantly binds to plasma protein fractions. Administered and endogenous GIP that binds to plasma proteins might lose its biological activity, while unbound peptide may have a lower plasma concentration than expected in cases where it does not bind to plasma proteins. Thus, without understanding the binding capabilities of GIP to plasma proteins, an accurate evaluation of the effects of clinically administered and endogenously released GIP may be hampered. The aim of the present study was to identify GIP binding proteins in the human peripheral circulation.
Materials and Methods
Plasma sample collection
Blood samples from the healthy volunteers were collected into vacutainers containing Na2-EDTA (1.5 mg/mL), and then plasma was separated immediately in a refrigerated centrifuge and stored in aliquots at –30°C until processing.
Fluorescent labeling of peptides
Fifty μL of 10–2 M fluorescein-5(6)-isothiocyanate (FITC, Sigma-Aldrich, MO, USA) dissolved in dimethylformamide was mixed with 50 μL of 10–4 M GIP, GLP-1 or glucagon (Peptide Institute, Osaka, Japan) and, then 150 μL of Na2CO3/NaHCO3 (0.1 M, pH 9.1) was added and continuously vortexed for 3 h in the dark at room temperature. The reaction mixture was then passed through a column prepacked with Sephadex G-25 Fine (GE Healthcare, Buckinghamshire, UK) saturated and eluted with 30 mM TES, 140 mM NaCl, 4 mM KCl (pH 7.4). Then the FITC labeled peptides were separated from unreacted FITC using the BioLogicTM LP System and Model 2110 Fraction Collector (Bio-Rad Laboratories, Hercules, CA, USA). Eluates were collected in fractions, each with a volume of 900 μL. The fraction with the highest fluorescence intensity determined at λex 485/λem 528 was used for subsequent experiments.
Production of antibodies and IgG purification
Synthetic peptides, [Cys0]-PEG-HKHNITQ, YAEGTF-PEG-[Cys0] and EGTFIS-PEG-[Cys0] were pretreated with a protein crosslinking and fixation reagent, mixed with respective untreated peptide, coupled to maleimide-activated mariculture keyhole limpet hemocyanin (Pierce), and immunized into Japanese white rabbits in the laboratory of Scrum Inc. (Tokyo, Japan) as described previously [8]. Polyclonal GIP antiserum and normal human IgG were purified using a MelonTM Gel IgG Purification Kit (Thermo Fisher Scientific, MA, USA) in accordance with the manufacturer’s instructions. Disulfide bonds of IgG were reduced by incubating with 100 mM dithiothreitol (Nakalai Tesque, Kyoto, Japan), 25 mM NH4HCO3 at 37°C for 60 min. Sandwich ELISA generated using the C-terminal specific anti-GIP antibody and N-terminal specific anti-active GIP antibody revealed highly specific for full-length GIP1–42.
Clear Native (CN)- and Blue Native (BN)-Polyacrylamide gel electrophoresis (PAGE)
For CN-PAGE of the plasma/peptide mixture, 6 μL of FITC-labeled peptide fraction was incubated with either 3 μL of human plasma, purified human IgG, albumin from human serum (10 mg/mL, Fujifilm Wako Pure Chemical, Osaka, Japan) or transferrin(apo) from human blood (5 mg/dL, Fujifilm Wako Pure Chemical) at 37°C for 18 h and diluted to 20 μL with Native PAGETM sample buffer (Thermo Fisher Scientific) and applied to a 15%–25% gradient gel (Perfect NT Gel; DRC, Osaka, Japan) using Native PAGETM Running Buffer (Thermo Fisher Scientific) and migrated at 120 V for 120 min. Fluorescently labeled HMW Native Marker Kit (GE Healthcare) and Native MarkTM Unstained Protein Standard (Thermo Fisher Scientific) was used as a marker of approximate molecular size. For BN-PAGE, the same buffers and running conditions were used, except that Native PAGETM Running Buffer was used as a positive electrode and Light Blue Cathode Buffer (Thermo Fisher Scientific) was used as a negative electrode. Fluorescence in the gel was visualized and photographed using the FAS-Digi Gel Documentation System (Nippon Genetics, Tokyo, Japan) and quantified by ImageJ software [9].
Western blotting
Western blotting was performed essentially as described [10, 11] except for the following modifications. After electrophoresis, the gel was placed in 20 mM Tris, 150 mM glycine, 0.1% SDS for 15 min, and transferred to an Immune-Blot® PVDF membrane (Trans-Blot® TurboTM Mini PVDF Transfer Packs, Bio-Rad Laboratories) using the Trans-Blot® TurboTM Transfer System at 25 V for 30 min. It was then incubated with 8% acetic acid for 15 min, sterilized water for 5 min, and air-dried to fix the protein. The PVDF membrane was then washed with sterilized water, blocked at 4°C overnight with 2% BSA/TBS containing 0.05% (w/v) Tween® 20 (TBS-T) and, after washing three times with TBS-T, probed with the C-terminal specific anti-GIP antibody diluted to 1:200 by Hikari A (Nacalai Tesque). After incubation with goat anti-rabbit IgG (1:3,000 dilution; Bio-Rad Laboratories) for 1 h, protein bands were detected with Amersham ECLTM Prime Western Blotting Detection Reagent (GE Healthcare), photographed by ImageQuant LAS 4000 (GE Healthcare) and analyzed by ImageJ software [9].
In-gel tryptic digestion and liquid chromatography tandem-mass spectrometry (LC-MS/MS)
Protein complexes separated by CN-PAGE were fragmented into peptides using a protocol for in-gel tryptic digestion as previously described [12] but with the following modifications. Fluorescent protein spots showing GIP-like immunoreactivity by western blotting in CN-PAGE were excised in rectangles of about 1–2 mm × 2 mm with a scalpel, washed with deionized water. Then the gel pieces were dehydrated in 200 μL acetonitrile for about 15 min and dried in an Iwaki VEC-100 micro centrifugal vacuum concentrator (Asahi Techno Glass Co., Chiba, Japan) for 60 min. The proteins in gel pieces were immersed into 100 μL of 10 mM DTT/25 mM ammonium bicarbonate for 1 h at 56°C, and, after washing with 100 μL of 25 mM ammonium bicarbonate for 10 min, incubated with 100 μL of 55 mM iodoacetamide in the dark at room temperature for 45 min. The pieces were washed with 25 mM ammonium bicarbonate for 10 min, dehydrated in 200 μL acetonitrile, dried in a centrifugal vacuum concentrator and then immersed in 20 μL of 50 mM Tris (pH 9.0) containing 100 ng/μL trypsin and 100 ng/μL Lys-C on ice for 45 min. Swollen gel pieces were placed in a minimal volume of 50 mM Tris (pH 9.0) at 37°C for 24 h, during which time peptide fragments digested in gel pieces diffused into the surrounding solution. The solution was transferred to siliconized plastic test tubes on ice. Peptide fragments remaining in gel pieces were further recovered after 20 min incubation at room temperature in minimal volumes of 5% (v/v) formic acid containing 50% (v/v) acetonitrile. The solutions containing peptides were pooled in siliconized tubes on ice. Combined solutions were desalted using stop and go extraction tips (stage tips) [13] filled with EmporeTM C18 sealant (3 M, MN, USA), prewashed with 50 μL of methanol, equilibrated with 50 μL of 70% acetonitrile containing 0.1% TFA, and then washed with 50 μL of 0.1% TFA. The peptides were eluted with 80 μL of 50% acetonitrile containing 0.1% TFA. The sample solutions were then lyophilized and re-dissolved in 20 μL of 3% acetonitrile containing 0.1% formic acid for LC-MS/MS analysis.
For the LC-MS/MS analysis, an ultra-sensitive LC-MS system combining LC (Easy-n LC 1000, Thermo Fisher Scientific) and Q-Exactive (Thermo Fisher Scientific) was used as described [14]. Database searches were performed using the SEQUEST algorithm incorporated into Proteome Discoverer 1.4.0.288 software (Thermo Fisher Scientific). The search parameters were as follows: enzyme, trypsin; variable modification, oxidation of Met residue; variable modification, carbamidomethylation of Cys residue; peptide ion mass tolerance, 6 ppm; fragment ion mass tolerance, 0.02 Da; and peptide charges, +2 to +8. The identified peptides were searched for in the decoy database and the false discovery rate was set as 0.01 using Percolator scoring with posterior error probability validation. Peptide quantitation was also performed using Proteome Discoverer 1.4.0.288.
In gel pronase E digestion and measurement of fluorescence
Human plasma pretreated for 1 h with or without the indicated doses of unlabeled GIP was incubated with FITC-GIP for 3 h at 37°C and applied and separated by CN-PAGE at 120 V for 2 h. Fluorescent bands that corresponded to albumin autofluorescence were excised in rectangles of about 1–2 mm × 2 mm with a scalpel under a FAS Digi Gel Imaging System (NIPPON Genetics) and washed with deionized water. Then the gel pieces were dehydrated in 100 μL acetonitrile for approximately 15 min and dried in an Iwaki VEC-100 micro centrifugal vacuum concentrator for 120 min. The dehydrated gel pieces were immersed into 490 μL of 0.2 mg/mL pronase E, 30 mM TES, 140 mM NaCl, 4 mM KCL, 10 mM CaCl2 (pH 7.5) and incubated at 37°C in the dark for 72 h. The solution was then transferred to polypropylene tubes and digested peptides remaining in gel pieces were further recovered by adding 25 μL of 50% acetonitrile for 10 min. Exudated fluids were combined into the polypropylene tubes. After the addition of 50 μL of 100 mM bicine, 100 mM NaCl (pH 8.5), fluorescent intensity was measured at λex/λem = 485/528 nm.
Results
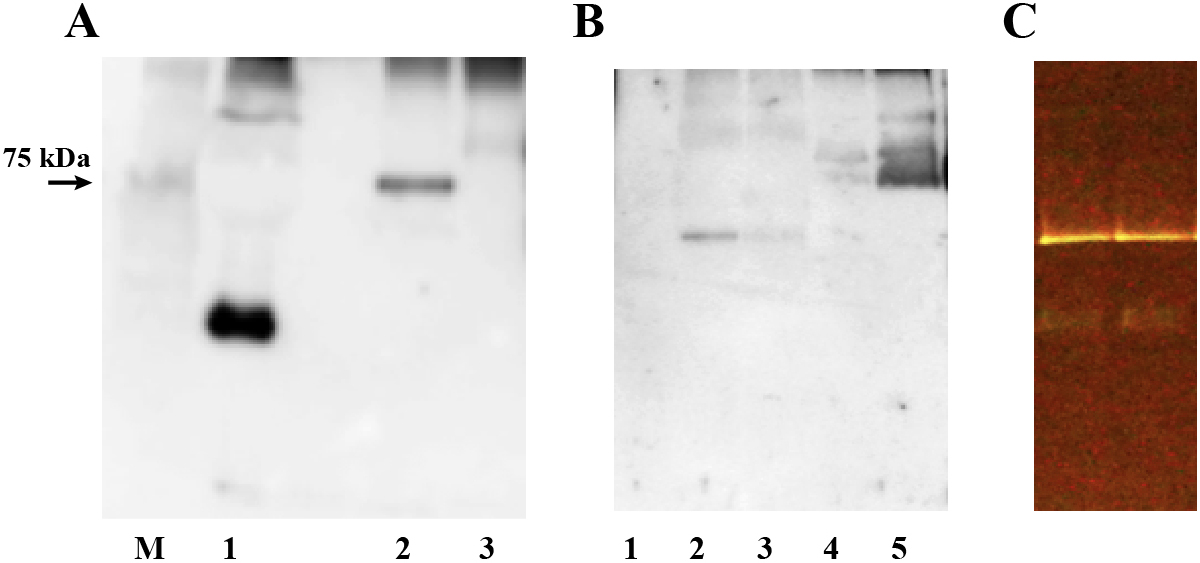
To isolate plasma proteins that bind to synthetic peptides, we incubated fresh human plasma with FITC-labeled GIP, GLP-1 and glucagon and subjected the reaction mixture to CN-PAGE. An autofluorescence band of 66 kDa was always detected when plasma or human serum albumin was applied to CN-PAGE even without the addition of any fluorescent peptide (Fig. 1A). However, addition of FITC-GIP to the plasma clearly intensified the fluorescence of this autofluorescence band and further generated an extra fluorescence band that migrated a longer distance on CN-PAGE (Fig. 1A, 1B). CN-PAGE of plasma incubated with FITC-GLP-1 or FITC-glucagon resulted in barely visible or negative fluorescence except for the autofluorescence band (Fig. 1B). Western blot analysis using anti-GIP antibody revealed the presence of GIP-like immunoreactivity exactly at the positions of the lower fluorescence-positive band (Fig. 1C). This GIP-positive/fluorescent band was excised and proteins in these gel sections were subsequently digested in-gel. The extracted peptides were analyzed by nano-flow reversed-phase LC-MS/MS to obtain peptide sequence identifications that were mapped to proteins in a sequence database. The database search revealed the presence of human serum albumin, IgG and transferrin in these in-gel digested proteins (Table 1). Considering the sequence coverage of the three identified proteins identified by LC-MS/MS analysis after in-gel tryptic digestion in the current analysis (Fig. 2A–2C), it is likely that all three full-length albumin, IgG and transferrin proteins comigrated with the fluorescent, GIP-positive protein complex.

Table 1
Results of database searches with the deduced amino acid sequences
| Protein Name |
Gene Name |
UniProt accession |
Score |
Coverage |
Unique Peptides |
PSMs |
MW [kDa] |
| Serotransferrin |
TF |
P02787 |
49.71 |
23.07 |
14 |
17 |
77.0 |
| Immunoglobulin heavy constant gamma 1 |
IGHG1 |
P01857 |
51.56 |
30.91 |
5 |
16 |
36.1 |
| Serum albumin |
ALB |
P02768 |
41.55 |
16.58 |
11 |
13 |
69.3 |
| Immunoglobulin heavy constant gamma 2 |
IGHG2 |
P01859 |
36.45 |
23.93 |
3 |
13 |
35.9 |
| Immunoglobulin heavy constant mu |
IGHM |
P01871 |
30.10 |
21.19 |
8 |
11 |
49.4 |
| Protein AMBP |
AMBP |
P02760 |
28.36 |
23.58 |
6 |
9 |
39.0 |
| Immunoglobulin kappa constant |
IGKC |
P01834 |
25.24 |
71.96 |
7 |
8 |
11.8 |
| Apolipoprotein C-III |
APOC3 |
P02656 |
19.72 |
51.52 |
4 |
6 |
10.8 |
| Isoform 2 of Fibrinogen alpha chain |
FGA |
P02671-2 |
23.65 |
10.9 |
5 |
6 |
69.7 |
| Transthyretin |
TTR |
P02766 |
18.10 |
38.10 |
4 |
5 |
15.9 |
Tandem mass (MS/MS) spectra of the peptide fragments were analyzed with mass spectrum analysis software, and the experimental MS/MS spectra of the peptides were compared with theoretical spectra computed from amino acid sequence data to identify the protein from which the digested peptides were derived. Candidates for complex formation proteins were listed from Coverage (%) and peptide spectrum matches (PSMs). From all proteins detected, only those with high PSM values are shown as candidates.

We determined whether GIP independently bound to any of the three proteins identified by LC-MS/MS analysis. FITC-GIP was incubated for 18 h with human serum albumin, purified normal human IgG or human transferrin proteins, and subjected to CN-PAGE or BN-PAGE. Immunoblotting with anti-C-terminal GIP antibody revealed the presence of GIP positive protein bands in the reaction with human serum albumin, purified human IgG and human transferrin (Fig. 3). Antibodies raised against active and inactive GIP N-terminal end sequences did not recognize proteins-bound GIP. These results demonstrate that GIP independently binds to each of these three proteins.
To confirm the binding of FITC-GIP to plasma proteins, we used CN-PAGE to quantify the fluorescence of FITC-GIP-bound proteins in the gel. Bindings of FITC-GIP to two plasma protein complexes were clearly detectable by fluorescence detection as early as 5–15 min and were increased at least during the initial 6 h (Fig. 4A). Quantification of the fluorescence of each protein band revealed that fluorescence of the two proteins intensified in a time-dependent fashion (approximately 6 h) (Fig. 4B, 4C). Because fluorescence elicited by FITC is often significantly reduced when attached to peptides or proteins [15], we circumvented this quenching phenomena by extensively proteolyzing FITC-bound GIP peptide protein complexes using in-gel digestion of bound proteins with pronase E to more accurately assess the magnitude of GIP binding to plasma proteins. The addition of 10–6 M FITC-GIP to human plasma and subsequent proteolysis resulted in markedly enhanced fluorescence signals while the pretreatment of plasma with excess unlabeled GIP dose-dependently suppressed the binding of FITC-GIP to plasma proteins (Fig. 4D). FITC-GIP peptide bound to human plasma proteins in a time-dependent manner and reached plateau after 24 hours (Fig. 4E). Taken together, these results demonstrate that GIP readily and significantly binds to plasma protein complexes containing albumin, IgG or transferrin.

Discussion
The present study revealed that FITC-GIP independently bound to albumin, IgG or transferrin and that FITC-GIP appeared to bind to plasma protein complexes composed of these proteins in the human peripheral circulation. Albumin, IgG or transferrin are highly abundant plasma components and GIP significantly bound to these proteins in a time-dependent manner. Thus, it is reasonable to assume that significant amounts of GIP peptide endogenously released or exogenously administered may undergo binding to such large molecular weight plasma proteins to form protein complexes in the human peripheral circulation.
In the present study, we developed a novel methodology to visualize plasma proteins that bind to GIP using CN-PAGE, which were then identified by subsequent in-gel trypsin digestion and LC-MS/MS. This protocol successfully isolated plasma proteins co-migrating with GIP and ultimately revealed that human serum albumin, IgG and serotransferrin are GIP-binding proteins. Unlike conventional SDS-PAGE, CN-PAGE can be performed under mild conditions and has been used to retain the supramolecular assemblies of plasma protein complexes that were dissociated under the harsher conditions of SDS-PAGE [16, 17]. In the binding experiment of GIP to human serum albumin and transferrin, we employed BN-PAGE in which the negatively charged Coomassie-dye in the presence of a neutral detergent mimicked the properties of anionic detergents, causing some dissociation, but allowing the easier electrophoresis of proteins complexes [16, 17]. However, GIP bound to human serum albumin and transferrin was not dissociated and was successfully detected under BN-PAGE. Binding proteins for GIP that have not been detected by SDS-PAGE were successfully identified using CN-PAGE and subsequent LC-MS/MS analysis, indicating it is a promising tool for the identification of plasma binding proteins for bioactive peptides.
To visualize GIP binding to plasma proteins, we used fluorescently labeled peptides and detected fluorescence using a fluorescence imaging system. Such protocols are simple and can be easily performed, but fluorescence-based assays have a serious drawback related to fluorescence quenching that accompanies the binding of fluorescently-labeled peptides to their binding proteins. Fortunately, in the present study, the fluorescence of FITC-GIP bound to plasma proteins was successfully determined because of the magnitude of the GIP binding capability to the plasma proteins. Immunoblotting using antibody against C-terminal end sequence of GIP confirmed the presence of GIP at the site of fluorescent positive protein complexes and clearly demonstrated that FITC-GIP bound to human serum albumin, IgG and transferrin. It should be noted that N-terminus antibodies for GIP did not recognize proteins-bound GIP, suggesting that GIP-binding proteins may have modified antigenicity of N-terminal sequences of GIP peptide. It is tempting to speculate that N-terminal sequence of GIP may be occupied by larger molecular weight proteins, such as albumin, IgG and transferrin, and that protein-bound GIP is resistant to DPP-4 inactivation.
In conclusion, we developed a novel methodology to identify plasma binding proteins for bioactive peptides and successfully identified serum albumin, IgG and transferrin as independent binding proteins for GIP. Our new protocol employs CN-PAGE and subsequent LC-MS/MS analysis of in-gel digests of fluorescently and/or immunologically detectable protein bands. The protein information can be used to identify reliable candidates for potential binding proteins.
Acknowledgments
The authors are grateful to Yukiko Kato for her technical assistance. This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (18H05383) to M.S., by Kitasato University ‘Shogaku-Kifu’ unrestricted research support to M.S. and by a grant from the Parents’ Association of Kitasato University School of Medicine to A.H. The funders had no role in the study design, data collection or analysis, decision to publish, or preparation of the manuscript. No additional external funding was received for this study. We thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Contributors
A.H. processed plasma samples and performed binding experiments. A.H., K.F., S.S., and A.M. performed CN- and BN-PAGE analyses and in-gel trypsin digests. R.K. and Y.K. performed LC-MS/MS analysis of excised protein samples. M.S. raised GIP antibodies, designed the entire study, analyzed data and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Disclosure
None of the authors have any potential conflicts of interest associated with this research.
References
- 1 Deacon CF, Ahren B (2011) Physiology of incretins in health and disease. The review of diabetic studies. Rev Diabet Stud 8: 293–306.
- 2 Christensen M, Vedtofte L, Holst JJ, Vilsboll T, Knop FK (2011) Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes 60: 3103–3109.
- 3 Dupre J, Ross SA, Watson D, Brown JC (1973) Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab 37: 826–828.
- 4 Cho YM, Kieffer TJ (2010) K-cells and glucose-dependent insulinotropic polypeptide in health and disease. Vitam Horm 84: 111–150.
- 5 Elahi D, McAloon-Dyke M, Fukagawa NK, Meneilly GS, Sclater AL, et al. (1994) The insulinotropic actions of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (7–37) in normal and diabetic subjects. Regul Pept 51: 63–74.
- 6 Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, et al. (1993) Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 91: 301–307.
- 7 Calanna S, Christensen M, Holst JJ, Laferrere B, Gluud LL, et al. (2013) Secretion of glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes: systematic review and meta-analysis of clinical studies. Diabetes Care 36: 3346–3352.
- 8 Fujimoto K, Hayashi A, Kodera Y, Saito T, Toki T, et al. (2017) Identification and quantification of plasma free salusin-β, an endogenous parasympathomimetic peptide. Sci Rep 7: 8275.
- 9 Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675.
- 10 Takada T, Kodera Y, Matsubara M, Kawashima Y, Maeda T, et al. (2013) Serum monomeric α2-macroglobulin as a clinical biomarker in diabetes. Atherosclerosis 228: 270–276.
- 11 Tani Y, Yamada S, Inoshita N, Hirata Y, Shichiri M (2015) Regulation of growth hormone secretion by (pro)renin receptor. Sci Rep 5: 10878.
- 12 Satoh M, Haruta-Satoh E, Omori A, Oh-Ishi M, Kodera Y, et al. (2005) Effect of thyroxine on abnormal pancreatic proteomes of the hypothyroid rdw rat. Proteomics 5: 1113–1124.
- 13 Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75: 663–670.
- 14 Suzuki S, Kodera Y, Saito T, Fujimoto K, Momozono A, et al. (2016) Methionine sulfoxides in serum proteins as potential clinical biomarkers of oxidative stress. Sci Rep 6: 38299.
- 15 Breen CJ, Raverdeau M, Voorheis HP (2016) Development of a quantitative fluorescence-based ligand-binding assay. Sci Rep 6: 25769.
- 16 Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, et al. (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem 278: 52873–52880.
- 17 Wittig I, Schagger H (2005) Advantages and limitations of clear-native PAGE. Proteomics 5: 4338–4346.
