Abstract
The cyclooxygenase2 (COX-2) enzyme catalyzes the first step of prostanoid biosynthesis, and is known for its crucial role in the pathogenesis of several inflammatory diseases including type 2 diabetes mellitus (T2DM). Although a variety of studies revealed that COX-2 played a role in the IL-1β induced β cell dysfunction, the molecular mechanism remains unclear. Here, using a cDNA microarray and in silico analysis, we demonstrated that inflammatory responses were upregulated in human T2DM islets compared with non-diabetic (ND) islets. COX-2 expression was significantly enhanced in human T2DM islets, correlated with the high inflammation level. PGE2, the catalytic product of COX-2, downregulated the functional gene expression of PDX1, NKX6.1, and MAFA and blunted the glucose induced insulin secretion of human islets. Conversely, inhibition of COX-2 activity by a pharmaceutical inhibitor prevented the β-cell dysfunction induced by IL-1β. COX-2 inhibitor also abrogated the IL-1β autostimulation in β cells, which further resulted in reduced COX-2 expression in β cells. Together, our results revealed that COX-2/PGE2 signaling was involved in the regulation of IL-1β autostimulation, thus forming an IL-1β/COX-2/PGE2 pathway loop, which may result in the high inflammation level in human T2DM islets and the inflammatory impairment of β cells. Breaking this IL-1β/COX-2/PGE2 pathway loop provides a potential therapeutic strategy to improve β cell function in the treatment of T2DM patients.
MULTIPLE STUDIES proved that islet inflammation can lead to β cell dysfunction and contributes to type 2 diabetes mellitus (T2DM) pathology. Glucotoxicity [1], lipotoxity [2, 3], immune cell infiltration [4-6], amyloid deposition [4, 7, 8], all these factors can lead to islet inflammation which is involved in β cell dysfunction.
Cyclooxygenase (COX), also known as prostaglandin-endoperoxide synthase (PTGS), the enzyme which is in charge of conversion of arachidonic acid into prostanoids, exists as two isoforms. COX-1 is a constitutive form which plays a role in prostaglandin-mediated physiological activity, whereas COX-2 is a regulated form which is thought to be induced by various stimuli including cytokines [9]. Decreased level of prostaglandin E2 (PGE2), the main catalytic product of COX-2, can improve glycemic control in obese people [10]. PGE2 can attenuate glucose-induced insulin secretion of β cells [11]. These studies indicated that COX-2 may play a pivotal role in the pathogenesis of T2DM, yet further research is still necessary to investigate the mechanism of its contribution to islet inflammation in T2DM.
In this study, we validated the up-regulation of COX-2 in human T2DM islets. Furthermore, we found that COX-2 was involved in the IL-1β autostimulation and IL-1β/COX-2/PGE2 pathway loop can lead to β cell functional impairments through depressing the expressions of β cell functional genes PDX1, NKX6.1 and MAFA. These data suggested that disturbing the IL-1β/COX-2/PGE2 pathway loop may be helpful for T2DM treatment.
Materials and Methods
Patient characteristics
All protocols were approved by the Medical Ethical Committee of Tianjin First Central Hospital (No. 2018N129KY). Pancreases were obtained from organ donors with or without T2DM disease and with research consents. The information of organ donors was shown in Table 1.
Table 1
The main information of organ donors
| Donor Characteristics |
Age (y) |
BMI (kg/m2) |
HbA1c (%) |
Male (%) |
Female (%) |
| ND (n = 16) |
47.31 ± 2.644 |
26.14 ± 1.124 |
5.578 ± 0.1152 |
85.7% |
14.2% |
| T2DM (n = 7) |
51.43 ± 3.046 |
25.58 ± 1.921 |
6.756 ± 0.5876 |
100% |
0% |
| p value |
0.7094 |
0.7499 |
0.0001 |
|
|
Data were shown as mean ± SEM.
Human islets were prepared by Collagenase NB1 (SERVA, Heidelberg, Germany) and Neutral Protease NB (SERVA, Heidelberg, Germany) digestion and continuous density purification. High purity islets (>80%) were collected and cultured in CMRL-1066 medium (Corning, Manassas, VA, USA), supplemented with 10% Human Serum Albumin (Baxter, Vienna, Austria), 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C in 5% CO2.
The human islets were incubated with 1 μM PGE2 (Sigma-Aldrich, Missouri, USA) or 10 ng/mL recombinant human IL-1β (Peprotech, New Jersey, USA) or a combination of 10 ng/mL recombinant human IL-1β and 10 μM Celecoxib (Selleck, Houston, Texas, USA) for 24 h.
Total RNA isolation, reverse transcription and real-time PCR
Total RNA was extracted from tissue samples or cultured cells using Trizol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer. Total RNA was reverse transcribed to cDNA using a RT (reverse transcriptase) reaction kit (Takara, Kohoku-cho, Kusatsu, Japan). Real-time PCR was performed using FS Essential DNA Green Master (Roche, Basel, Switzerland). The primers used to assess the expression of mRNA were shown in Supplementary Table 1.
Cell culture and treatments
Mouse pancreatic β cell line NIT-1 was grown in DMEM/F12 medium supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. The cells were incubated at 37°C with 5% CO2.
The cells were incubated with 1 μM PGE2 (Sigma-Aldrich, Missouri, USA) or 10 ng/mL recombinant mouse IL-1β (Peprotech, New Jersey, USA) or a combination of 10 ng/mL recombinant mouse IL-1β and 10 μM Celecoxib (Selleck, Houston, Texas, USA) for 48 h.
Insulin secretion assay
10 islets were pretreated in 1 mL 1.67 mM low-glucose Krebs-Ringer bicarbonate buffer (KRB; supplemented with 0.5% BSA, pH 7.4) for 1 h in 12-well plate, then medium was removed, followed by sequential treatment with 1 mL low-glucose KRB solution (1.67 mM) for 1 h and high-glucose KRB solution (16.7 mM) for 1 h. The media at low and high glucose levels were collected separately. Insulin concentration was measured by ELISA (Human Insulin Elisa Kit; Mercodia, Uppsala, Sweden). Insulin secretion of islets was expressed as the glucose stimulated index (GSI; insulin content in the 16.7 mM glucose media/insulin content in the 1.67 mM glucose media) to assess glucose responsiveness.
Immunohistochemistry
Formalin-fixed, paraffin-embedded pancreas sections (3 μm) were incubated with COX-2 (1:100, Bioworld, Nanjing, China) primary antibodies, along with TRITC AffiniPure Goat Anti-Rabbit IgG H&L (1:100, Jackson Immunoresearch Laboratories and Molecular Probes, West Grove, PA, USA) secondary antibodies. Counterstaining was performed with DAPI (Vector, Burlingame, CA, USA). We used Pannoramic MIDI and Pannoramic Viewer (3DHistech, Budapest, Hungary) to scan stained slides and capture images.
Microarray
Human islet samples (>80% purity) were isolated from 2 human ND (H046 and H104) and 2 T2DM (HE003, HE005) organ donors.
Total RNA was extracted and purified (mirVana™ miRNA Isolation Kit; Cat.# AM1561, Ambion, Austin, TX, US), amplified and labeled (Low Input Quick Amp WT Labeling Kit; Cat.# 5190-2943, Agilent Technologies, Santa Clara, CA, US) following the manufacturer’s instructions. Labeled cRNA were purified by RNeasy mini kit (Cat.# 74106, QIAGEN, Dusseldorf, Germany). Each slide was hybridized with Cy3-labeled cRNA using a Gene Expression Hybridization Kit (Cat.# 5188-5242, Agilent Technologies, Santa Clara, CA, USA) and slides were washed in staining dishes (Cat.# 121, Thermo Scientific, Waltham, MA, USA) with a Gene Expression Wash Buffer Kit (Cat.# 5188-5327, Agilent Technologies, Santa Clara, CA, US), then scanned with an Agilent Microarray Scanner (Cat.# G2565CA, Agilent Technologies, Santa Clara, CA, US) with the default settings. The data were extracted with Feature Extraction software 10.7 (Agilent Technologies, Santa Clara, CA, US). The raw data were normalized using the Quantile algorithm of the limma package in R. A microarray analysis was performed by Shanghai Biotechnology Cooperation (Shanghai, China). The series number of our microarray data uploaded to Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) is GSE118139.
GSE38642 was downloaded from the GEO database which includes 54 ND and 9 T2DM islets samples. Differentially expressed mRNAs between T2DM and ND islets samples were analyzed with GEO2R software (http://www.ncbi.nlm.nih.gov/geo/geo2r/), p values <0.05 and |log2FoldChange| ≥1 were defined as the cut-off criteria.
Statistical analysis
All data were analyzed by two tailed student’s t test using GraphPad Prism Version 7.0d. p values of <0.05 were considered statistically significant.
Results
Islet inflammation and COX-2 were elevated in T2DM islets
By analyzing of our cDNA microarray (GSE118139), we found that the mRNA expressions of multiple cytokines (Fig. 1A) as well as inflammatory response (GO: 0006954) (Fig. 1B) were upregulated in human T2DM islets compared with ND islets. Then by analyzing GSE38642 downloaded from GEO database, we found COX-2 was among the top 20 differentially expressed genes in microarray (Fig. 1C). Furthermore, we confirmed the elevation of COX-2 expression in human T2DM islets (Fig. 1D). To further detect the expression of COX-2, immunofluorescence staining was performed in human ND and T2DM pancreas sections (Fig. 1E). Surprisingly, we found that COX-2 staining was markedly increased in the human T2DM islets compared with ND islets. These data indicate that COX-2 is highly expressed in human T2DM islets, which is associated with the high inflammation level of the islets.
PGE2, the catalytic product of COX-2, impaired the function of β cells
It is well known that COX-2 plays a key role in PGE2 biosynthesis, which is related to proinflammatory reactions [12-14]. To determine the function of COX-2 in β cells, we first examined the effect of PGE2, the classical product of COX-2, on the β cell function. As shown in Fig. 2A, the expressions of β cell functional genes PDX1, NKX6.1, MAFA were significantly down-regulated with PGE2 treatment. We also confirmed the results using human ND islets (Fig. 2B). To further validate the PGE2 function, we assessed glucose stimulated index (GSI) of human ND islets under PGE2 treatment. As shown in Fig. 2C, the GSI was significantly decreased after the PGE2 treatment. This result indicated that PGE2 can impair glucose induced insulin secretion of islets. Taken together, these results imply that PGE2 can impair the function of β cells.
COX-2 was involved in IL-1β autostimulation
As IL-1β is an important mediator of inflammatory response and is closely related to T2DM [15] which is also confirmed by our microarray results, we treated the NIT-1 with IL-1β and examined the expression of COX-2. As expected, the COX-2 gene expression was strikingly up-regulated by IL-1β (Fig. 3A). Interestingly, we found the event could be abrogated by Celecoxib, a selective inhibitor of COX-2 activity (Fig. 3A). These results also confirmed by using human ND islets (Fig. 3B). These results indicated that Celecoxib can not only inhibit the activity of COX-2 but also inhibit the gene expression of COX-2.
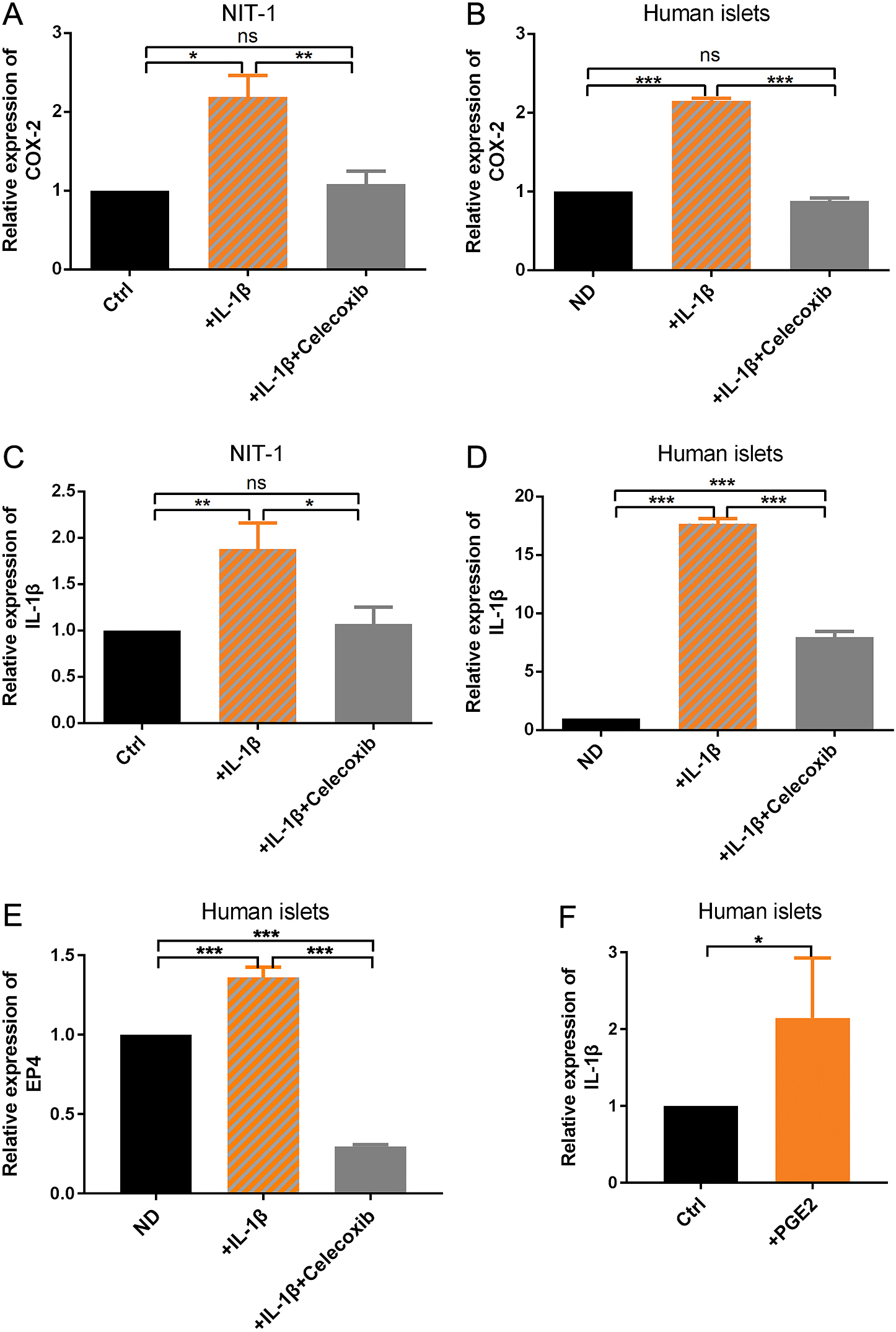
It has been proposed that there is an IL-1β “autostimulation” in β cells: IL-1β expression can be further enhanced by itself through a positive feedback loop of IL-1β signaling [16]. So we hypothesized that Celecoxib inhibited the gene expression of COX-2 by inhibiting the IL-1β autostimulation. In order to identify the relation of COX-2 and IL-1β, we detected the mRNA expression of IL-1β itself under IL-1β or IL-1β combined with Celecoxib treatment. Results showed that IL-1β treatment can significantly increase the mRNA expression of IL-1β itself (Fig. 3C), consistent with the notion of “autostimulation”. As expected, Celecoxib abrogated the IL-1β autostimulation, as indicated by the significantly decreased IL-1β expression (Fig. 3C). These results were also verified by using human ND islets (Fig. 3D). Furthermore, the gene expression of E-Prostanoid Receptor4 (EP4), a receptor of PGE2, was significantly higher in IL-1β-treated human islets which was blocked by Celecoxib (Fig. 3E). The increased expression of EP4 indicates the increased production of PGE2. In parallel, PGE2 treatment boosted the IL-1β expression in human ND islets (Fig. 3F), which further confirmed that COX-2 is a key mediator regulating IL-1β autostimulation and there exists an IL-1β/COX-2/PGE2 feedback loop in inflammatory islets.
IL-1β/COX-2/PGE2 pathway loop led to β cell dysfunction
To determine the effect of IL-1β/COX-2/PGE2 pathway loop on β cell function, we examined the expression of β cell functional genes under IL-1β or IL-1β combined with Celecoxib treatment. Just as expected, the expressions of β cell critical functional genes PDX1, NKX6.1 and MAFA were significantly downregulated by IL-1β and could be restored by Celecoxib both in NIT-1 cell line (Fig. 4A) and human ND islets (Fig. 4B). Taken together, these results suggest that IL-1β/COX-2/PGE2 pathway loop can result in β cell dysfunction (Fig. 4C).
Discussion
It was observed that T2DM islets exhibited features of inflammation, including increased expression of cytokines [17] and immune cell infiltration [18, 19], which resulted in disruption the islets function and mass [20, 21]. Consistent with these results, our results of cDNA microarray also confirmed the existence of islet inflammation in T2DM islets (Fig. 1A and 1B). IL-1β, either derived from macrophages [22] or islet cells [1, 23], is a critical proinflammatory mediator of β cell impairment in T2DM. In the results of our microarray, we also found that IL-1β were enriched in human T2DM islets (Fig. 1A), which once again confirmed the important role of IL-1β in T2DM. In this study, we provided solid evidence for the abnormal expression of COX-2 in the islets of organ donors with T2DM (Fig. 1D and 1E). This is consistent with previous studies reporting the high expression of COX-2 in animal models with T2DM [10]. Tran et al. [24] demonstrated that IL-1β induced COX-2 gene expression in β cells. Here, we identified the association of COX-2 expression with islet inflammation in human T2DM islets. The abnormal COX-2 expression is possibly caused by highly expressed proinflammatory cytokines in islets and may be accounted, at least partially, for the β cell dysfunction in human T2DM islets.
PGE2 is the most known catalytic product of COX-2 [25] which has 4 receptors, namely E-Prostanoid Receptor (EP)1–4, transducting PGE2 signaling. Among them, EP3 and EP4 are expressed both in human α and β cells [26]. Both PGE2 production and EP3 expression were increased in T2DM islets [27]. Watanabe et al. [28] proved that IL-1β stimulated the EP4 expression in human chondrocytes by increasing the production of PGE2. In our study, we also found that IL-1β can stimulate the gene expression of EP4 in human islets (Fig. 3E) which indicates the increased production of PGE2. Even previous studies have primarily concluded that PGE2 impairs insulin secretion of β cells [11, 27, 29, 30], few study has paid attention to the molecular changes of β cells under PGE2 treatment. Here, we identified that PGE2 can significantly suppress the expressions of β cell functional genes PDX1, MAFA, NKX6.1, both in NIT-1 cell line (Fig. 2A) and human ND islets (Fig. 2B) and complemented the molecular mechanism of PGE2’s effect on β cells.
Previous studies have demonstrated that IL-1β could induce COX-2 expression, PGE2 synthesis and EP3 expression in β cells [24]. Here, by using NIT-1 cell line as well as human ND islets, we also confirmed the regulation of COX-2 by IL-1β (Fig. 3A and 3B). Besides, we confirmed the upregulation of EP4 by IL-1β in human islets, which may be one of the routes of islet dysfunction due to IL-1β (Fig. 3E). IL-1β can enhance itself synthesis through a feedback loop of IL-1β signaling. This autostimulation can lead to accumulation of IL-1β and amplify inflammation [16, 31]. Surprisingly, in our study, we found that COX-2 selective inhibitor, Celecoxib, could suppress the autostimulation of IL-1β both in β cells (Fig. 3C) and human islets (Fig. 3D). So we speculated that COX-2 can regulate IL-1β expression by taking part in IL-1β autostimulation. Furthermore, in our study, we proved that IL-1β/COX-2/PGE2 pathway loop could destruct β cell function by down-regulating PDX1, NKX6.1 and MAFA (Fig. 4A and 4B). Celecoxib is a COX-2 selective inhibitor which has anti-inflammatory and anti-cancer functions [32, 33]. Here, we found that Celecoxib could prevent IL-1β’s adverse effects on β cell function, even though partially (Fig. 4A and 4B). These results confirmed the destructive effects of COX-2 on β cells. What is more, Celecoxib could repair the dysfunction of β cells by inhibiting IL-1β/COX-2/PGE2 pathway loop. The results hinted that COX-2 inhibitors may be a potential medication for T2DM therapy.
Collectively, our study demonstrated the abnormal expression of COX-2 in human T2DM islets and established its unique role both in β cell dysfunction and IL-1β autostimulation. This IL-1β/COX2/PGE2 pathway loop might be responsible for the high inflammation and β cell dysfunction in T2DM islets. The insight gained from this study have supplemented the understanding of the molecular mechanisms underlying β cell dysfunction in inflammatory islets, and might lead to the development of treatment of T2DM.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81870535), National key research and development program (2016YFC1305104), Key projects of Tianjin Natural Science Foundation (18JCZDJC33100), Tianjin Clinical Research Center for Organ Transplantation Project (15ZXLCSY00070), and Foundation of State Key Laboratory of Medicinal Chemical Biology (Nankai University) (2018016).
Disclosure
The authors declare that they have no conflict of interest to disclose.
Reference
- 1 Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, et al. (2002) Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110: 851–860.
- 2 Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, et al. (2012) Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab 15: 518–533.
- 3 Mukhuty A, Fouzder C, Mukherjee S, Malick C, Mukhopadhyay S, et al. (2017) Palmitate induced fetuin-A secretion from pancreatic beta-cells adversely affects its function and elicits inflammation. Biochem Biophys Res Commun 491: 1118–1124.
- 4 Kamata K, Mizukami H, Inaba W, Tsuboi K, Tateishi Y, et al. (2014) Islet amyloid with macrophage migration correlates with augmented beta-cell deficits in type 2 diabetic patients. Amyloid 21: 191–201.
- 5 Butcher MJ, Hallinger D, Garcia E, Machida Y, Chakrabarti S, et al. (2014) Association of proinflammatory cytokines and islet resident leucocytes with islet dysfunction in type 2 diabetes. Diabetologia 57: 491–501.
- 6 Martino L, Masini M, Bugliani M, Marselli L, Suleiman M, et al. (2015) Mast cells infiltrate pancreatic islets in human type 1 diabetes. Diabetologia 58: 2554–2562.
- 7 Zhao HL, Lai FM, Tong PC, Zhong DR, Yang D, et al. (2003) Prevalence and clinicopathological characteristics of islet amyloid in chinese patients with type 2 diabetes. Diabetes 52: 2759–2766.
- 8 Montane J, de Pablo S, Castano C, Rodriguez-Comas J, Cadavez L, et al. (2017) Amyloid-induced beta-cell dysfunction and islet inflammation are ameliorated by 4-phenylbutyrate (PBA) treatment. FASEB J 31: 5296–5306.
- 9 Robertson RP (1998) Dominance of cyclooxygenase-2 in the regulation of pancreatic islet prostaglandin synthesis. Diabetes 47: 1379–1383.
- 10 Neuman JC, Schaid MD, Brill AL, Fenske RJ, Kibbe CR, et al. (2017) Enriching islet phospholipids with eicosapentaenoic acid reduces prostaglandin e2 signaling and enhances diabetic beta-cell function. Diabetes 66: 1572–1585.
- 11 Tran PO, Gleason CE, Poitout V, Robertson RP (1999) Prostaglandin E(2) mediates inhibition of insulin secretion by interleukin-1beta. J Biol Chem 274: 31245–31248.
- 12 Wang M, Wang Y, Xie T, Zhan P, Zou J, et al. (2019) Prostaglandin E2/EP2 receptor signalling pathway promotes diabetic retinopathy in a rat model of diabetes. Diabetologia 62: 335–348.
- 13 Sheppe AEF, Kummari E, Walker A, Richards A, Hui WW, et al. (2018) PGE2 augments inflammasome activation and M1 polarization in macrophages infected with Salmonella Typhimurium and Yersinia enterocolitica. Front Microbiol 9: 2447.
- 14 Arima K, Ohmuraya M, Miyake K, Koiwa M, Uchihara T, et al. (2019) Inhibition of 15-PGDH causes Kras-driven tumor expansion through prostaglandin E2-ALDH1 signaling in the pancreas. Oncogene 38: 1211–1224.
- 15 Zhao G, Dharmadhikari G, Maedler K, Meyer-Hermann M (2014) Possible role of interleukin-1beta in type 2 diabetes onset and implications for anti-inflammatory therapy strategies. PLoS Comput Biol 10: e1003798.
- 16 Boni-Schnetzler M, Boller S, Debray S, Bouzakri K, Meier DT, et al. (2009) Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 150: 5218–5229.
- 17 Donath MY, Boni-Schnetzler M, Ellingsgaard H, Halban PA, Ehses JA (2010) Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends Endocrinol Metab 21: 261–267.
- 18 Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, et al. (2007) Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56: 2356–2370.
- 19 Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG (2009) Islet-associated macrophages in type 2 diabetes. Diabetologia 52: 1686–1688.
- 20 Cnop M, Abdulkarim B, Bottu G, Cunha DA, Igoillo-Esteve M, et al. (2014) RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 63: 1978–1993.
- 21 Eizirik DL, Miani M, Cardozo AK (2013) Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 56: 234–241.
- 22 Cavelti-Weder C, Timper K, Seelig E, Keller C, Osranek M, et al. (2016) Development of an interleukin-1beta vaccine in patients with type 2 diabetes. Mol Ther 24: 1003–1012.
- 23 Anquetil F, Sabouri S, Thivolet C, Rodriguez-Calvo T, Zapardiel-Gonzalo J, et al. (2017) Alpha cells, the main source of IL-1beta in human pancreas. J Autoimmun 81: 68–73.
- 24 Tran PO, Gleason CE, Robertson RP (2002) Inhibition of interleukin-1beta-induced COX-2 and EP3 gene expression by sodium salicylate enhances pancreatic islet beta-cell function. Diabetes 51: 1772–1778.
- 25 Robertson RP (2017) The COX-2/PGE2/EP3/Gi/o/cAMP/GSIS pathway in the islet: the beat goes on. Diabetes 66: 1464–1466.
- 26 Bramswig NC, Everett LJ, Schug J, Dorrell C, Liu C, et al. (2013) Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. J Clin Invest 123: 1275–1284.
- 27 Kimple ME, Keller MP, Rabaglia MR, Pasker RL, Neuman JC, et al. (2013) Prostaglandin E2 receptor, EP3, is induced in diabetic islets and negatively regulates glucose- and hormone-stimulated insulin secretion. Diabetes 62: 1904–1912.
- 28 Watanabe Y, Namba A, Honda K, Aida Y, Matsumura H, et al. (2009) IL-1beta stimulates the expression of prostaglandin receptor EP4 in human chondrocytes by increasing production of prostaglandin E2. Connect Tissue Res 50: 186–193.
- 29 Meng Z, Lv J, Luo Y, Lin Y, Zhu Y, et al. (2009) Forkhead box O1/pancreatic and duodenal homeobox 1 intracellular translocation is regulated by c-Jun N-terminal kinase and involved in prostaglandin E2-induced pancreatic beta-cell dysfunction. Endocrinology 150: 5284–5293.
- 30 Oshima H, Taketo MM, Oshima M (2006) Destruction of pancreatic beta-cells by transgenic induction of prostaglandin E2 in the islets. J Biol Chem 281: 29330–29336.
- 31 Wu L, Guo F, Wu Y, Wang Q, Ma X, et al. (2017) The role of FoxO1 in interleukin-1beta-induced autostimulation in retina endothelial cells and retinas of diabetic rats. Microvasc Res 112: 93–100.
- 32 Mhillaj E, Morgese MG, Tucci P, Furiano A, Luongo L, et al. (2018) Celecoxib prevents cognitive impairment and neuroinflammation in soluble amyloid beta-treated rats. Neuroscience 372: 58–73.
- 33 Qiu Z, Zhang C, Zhou J, Hu J, Sheng L, et al. (2019) Celecoxib alleviates AKT/c-Met-triggered rapid hepatocarcinogenesis by suppressing a novel COX-2/AKT/FASN cascade. Mol Carcinog 58: 31–41.



