Abstract
The fat-specific protein 27 gene (Fsp27) belongs to the cell death-inducing DNA fragmentation factor 45-like effector family. Fsp27 is highly expressed in adipose tissue and fatty liver. In adipocytes, FSP27 localizes to the membrane of lipid droplets and promotes lipid droplet hypertrophy. Recently, FSP27 was shown to consist of two isoforms, FSP27α and FSP27β. Previously, we demonstrated that Fsp27a is directly regulated by peroxisome proliferator-activated receptor γ (PPARγ) in fatty livers of genetically obese leptin deficient ob/ob mice and that Fsp27b may potentially be regulated by different factors transcriptionally as they both have a different promoter region. Thus, the aim of the present study was to elucidate whether Fsp27b is regulated by PPARγ in fatty liver. Fsp27a and Fsp27b were markedly induced in fatty liver of ob/ob mice compared with those in the normal liver. However, both Fsp27a/b were expressed at markedly lower levels in liver-specific PPARγ knockout mice with an ob/ob background. Further, the PPAR response element (PPRE) for the PPARγ-dependent promotion of Fsp27b promotor activity was revealed at position –1,163/–1,151 from the transcriptional start site (+1). Interestingly, the cis-element responsible for the PPARγ-dependent induction of Fsp27b was the same as that responsible for PPARγ-dependent induction of Fsp27a. These results suggest that PPARγ regulates not only Fsp27a but also Fsp27b in fatty liver of ob/ob mice through a common PPRE.
FAT-SPECIFIC PROTEIN 27 (FSP27) was initially identified from mouse adipocyte TA cell lines as a mature adipocyte-specific gene [1, 2] and is considered to belong to the protein family of cell death-inducing DNA fragmentation factor 45-like effector (CIDE) based on the homology of protein sequences [2]. The human homolog of mouse FSP27 was also reported as CIDEC [3]. FSP27 was highly expressed in white or brown adipose tissues and localized to the membrane of lipid droplets (LDs) in adipocytes [4] through amino acids 173–220 of the FSP27 protein [5]. In earlier adipocyte studies, FSP27 was shown to promote LD-LD fusion and to form enlarged and unilocular LDs in cooperation with perilipin1, another LD-associated protein [6]. Furthermore, conventional FSP27-null mice showed small white adipose tissue mass due to decreased triglyceride (TG) levels and also showed protection from diet-induced obesity and insulin resistance [7, 8]. Adipocyte-specific FSP27-null mice also had small white adipose tissue mass and hepatosteatosis [9]. These results reveal that FSP27 contributes to TG storage in white adipose tissue (WAT) by promoting the enlargement of unilocular LDs.
Peroxisome proliferator-activated receptor γ (PPARγ) is a nuclear receptor that heterodimerizes with the retinoid X receptor α (RXRα), another nuclear receptor that is activated by 9-cis retinoic acid and activates multiple genes. In earlier studies, we generated a liver-specific PPARγ-null mouse (ob/ob-PPARγLKO) with a genetic ob/ob background, which is a well-characterized model of type 2 diabetes, obesity, and fatty liver. The ob/ob-PPARγLKO mouse showed a marked decrease in hepatic TG content and an improvement of fatty liver compared to wild ob/ob mice (ob/ob-PPARγWT), indicating that PPARγ is capable of activating the expression of genes involved in TG accumulation in hepatocytes and promoting the generation of fatty liver [10]. Furthermore, we isolated Fsp27 by subtraction screening using mRNAs from ob/ob-PPARγWT and ob/ob-PPARγLKO livers and established it as a direct mediator of PPARγ-dependent fatty liver in ob/ob mice by increasing hepatic TG levels [11]. Hepatic FSP27 also plays a critical role in fasting- [12] and alcohol- [13] induced hepatosteatosis.
Recently, two FSP27 isoforms were identified: FSP27α and FSP27β. FSP27α and FSP27β have a different exon 1 (exons 1a and 1b) and the same exons 2–6. The translation start site of FSP27α exists in exon 2, whereas that of FSP27β exists in exon 1b. Therefore, FSP27β contains an additional 10 amino acids at the N-terminus. Although both isoforms have the same intracellular function of promoting the enlargement of LDs, FSP27β is more stable than FSP27α [14].
The transcriptional regulation of Fsp27a/b remains controversial. Fsp27b mRNA was shown to be markedly induced in fasted liver and fatty liver of ob/ob mice. Cyclic AMP-responsive element-binding protein H (CREBH), a transcription factor known to be highly expressed in the liver, was reported as a critical factor for Fsp27b mRNA induction in the fasting normal liver [14]. We previously demonstrated that Fsp27a is directly regulated by PPARγ in the fatty livers of ob/ob mice through a functional PPAR response element (PPRE) on the promotor region [11]. These results raise the question of whether PPARγ in fatty liver of ob/ob mice contributes to the transcriptional regulation of Fsp27b mRNA. In this study, we investigated the role of PPARγ in the regulation of Fsp27b expression in fatty liver.
Materials and Methods
Animal studies
This study was approved by the Center for Experimental Animals at Fukuoka University (Permission number: 1815119) and carried out in accordance with the guidelines of the Center. Liver-specific PPARγ knockout mice (PPARγLKO) were generated by breeding Pparg-floxed mice with mice expressing the Cre recombinase under the control of an albumin promoter. In addition, PPARγLKO were generated on ob/ob (ob/ob-PPARγLKO) and normal genetic backgrounds (OB/OB-PPARγLKO), as previously described [10].
For the fasting and refeeding study, C57BL/6JJc1 mice (male, 10 weeks) were fed a regular chow diet (MF, Oriental Yeast, Fukuoka, Japan) ad libitum until the experimental treatment was commenced. Mice in the fasting group (n = 3) were fasted for 24 h and then killed. Mice in the refeeding group (n = 3) were fasted for 24 h, refed with a 50% (w/w) sucrose/MF diet for 24 h, and then killed. Mice in the control group (n = 3) were fed ad libitum with a regular chow diet and euthanized at the same time as the refeeding group.
RNA extraction and quantitative real-time polymerase chain reaction
RNA was extracted using the TRIzol reagent (Thermo Fisher Scientific, MA, USA) and quantitative real-time polymerase chain reaction (QRT-PCR) was performed using cDNA generated using 1 μg of total RNA with an AffinityScript QRT-PCR cDNA Synthesis Kit (Thermo Fisher Scientific, MA, USA). The primer sequences used were as follows: Pparg: forward, 5'-CATGGCCATTGAGTGCCGAGT-3' and reverse, 5'-ACATCCCCACAGCAAGGCAC-3'; Fsp27a: forward, 5'-GCCACGCGGTATTGCCAGGA-3' and reverse, 5'-GGGTCTCCCGGCTGGGCTTA-3'; Fsp27b: forward, 5'-GTGACCACAGCTTGGGTCGGA-3' and reverse, 5'-GGGTCTCCCGGCTGGGCTTA-3'; adipocyte protein 2 (ap2): forward, 5'-GATGCCTTTGTGGGAACCTG-3' and reverse, 5'-GAATTCCACGCCCAGTTTGA-3'; Crebh: forward, 5'-GATCCGGAACAAACAGTCAGC-3' and reverse, 5'-GCAAAACCTTCCTCTGCAGCTC-3'; apolipoprotein A-IV (ApoA-IV): forward, 5'-GCTAAGCAACAATGCCAAGG-3' and reverse, 5'-CCACTCAGCTGTACGACAAAGG-3'; acidic ribosomal phosphoprotein P0 (36b4): forward, 5'-AAACTGCTGCCTCACATCCG-3' and reverse, 5'-TGGTGCCTCTGGAGATTTTCG-3'. QRT-PCR reactions were performed using the Brilliant III Ultra-Fast SYBR Green QRT-PCR Master Mix (Thermo Fisher Scientific, MA, USA) in an Mx3005P Real-Time PCR System (Thermo Fisher Scientific, MA, USA). Expression levels of mRNAs were normalized to those of 36b4 mRNA.
Construction of luciferase reporter plasmids
The transcription start site of mouse Fsp27b was determined previously [14]. The –2.7 kb (D1), –2.4 kb (D2), –1.9 kb (D3), –1.4 kb (D4), –0.5 kb (D5), and –0.2 kb (D6) fragments from the transcriptional start site (+1) of the mouse Fsp27b 5'-upstream region, containing CACC sites at the 5'-end of the primers, were amplified by PCR and cloned into the Gateway entry vector pENTR/D-TOPO (Thermo Fisher Scientific, MA, USA), and then recombined into the destination vector pGL4.17 (Promega, WS, USA), which was prepared using the Gateway Vector Conversion System according to the manufacturer’s instructions (Thermo Fisher Scientific, MA, USA). The forward primer sequences used were as follows: D1: 5'-CACCCTCCCATTGCTCATTCG-3'; D2: 5'-CACCATCAGCTGTGCCTACGGATG-3'; D3: 5'-CACCCTGTAGCCCAGGCTAGCT-3'; D4: 5'-CACCTGAGACAGGGCCAACTCT-3'; D5: 5'-CACCGAACCAGAGAACAGCACTACAGG-3'; D6: 5'- CACCCGTGGAGTTTCTGGACTATAGGTG-3'. The reverse primer, which was common for all constructs, was as follows: 5'-TGTTTCTCCGACCCAAGCTG-3'.
Production and infection of recombinant adenovirus
Recombinant adenoviruses expressing mouse PPARγ and Lac were constructed using the Adeno-X Expression System 2 Kit (Takara Bio USA, Inc., CA, USA) as described previously [11]. The adenovirus titer was determined using the Adeno-X Rapid Titer Kit (Takara Bio USA, Inc., CA, USA). AML-12 cells were infected at 8,000 multiplicity of infection (moi) of each adenovirus in serum-free DMEM/F12 medium for 1 h.
Statistical analysis
Quantitative values are presented as mean ± standard error of the mean (SEM). Statistical significance was determined using one-way ANOVA with the Tukey multiple comparison test, and defined at p < 0.05.
Results
Effect of fatty liver and hepatic PPARγ on Fsp27b expression
To elucidate the contribution of hepatic PPARγ to Fsp27a and Fsp27b expression in steatotic ob/ob liver, we performed QRT-PCR analyses using liver-specific PPARγ knockout mice with an ob/ob background (ob/ob-PPARγLKO). Although Pparg mRNA is induced on the ob/ob background, Pparg mRNA was expressed at a basal level in ob/ob-PPARγLKO (Fig. 1A) mice. Fsp27a mRNA expression levels in control ob/ob-PPARγLKO mice and the PPARγ-specific ligand rosiglitazone (RGZ)-treated ob/ob-PPARγLKO mice were approximately 54% and 70% that of ob/ob-PPARγWT. Expression of Fsp27a mRNA in control ob/ob-PPARγWT mice was induced approximately 3-fold by RGZ (Fig. 1B). It is noteworthy that Fsp27b mRNA expression in control ob/ob-PPARγLKO and the RGZ-treated ob/ob-PPARγLKO mice was approximately 7% and 5% that in ob/ob-PPARγWT mice. In addition, the expression of control Fsp27b mRNA in ob/ob-PPARγWT was induced approximately 1.3-fold without and with RGZ (Fig. 1C). However, there was no distinct difference in Crebh mRNAs between PPARγWT and PPARγLKO mice of the control or RGZ groups in the present study (Fig. 1D). Although the expression of Apo-AIV mRNA, which is a CREBH-target gene [15], in ob/ob genetic background mice was higher than that in normal OB/OB mice, its expression in ob/ob-PPARγLKO mice was slightly decreased (control) or increased (RGZ) as compared with that in ob/ob-PPARγWT mice (Fig. 1E).
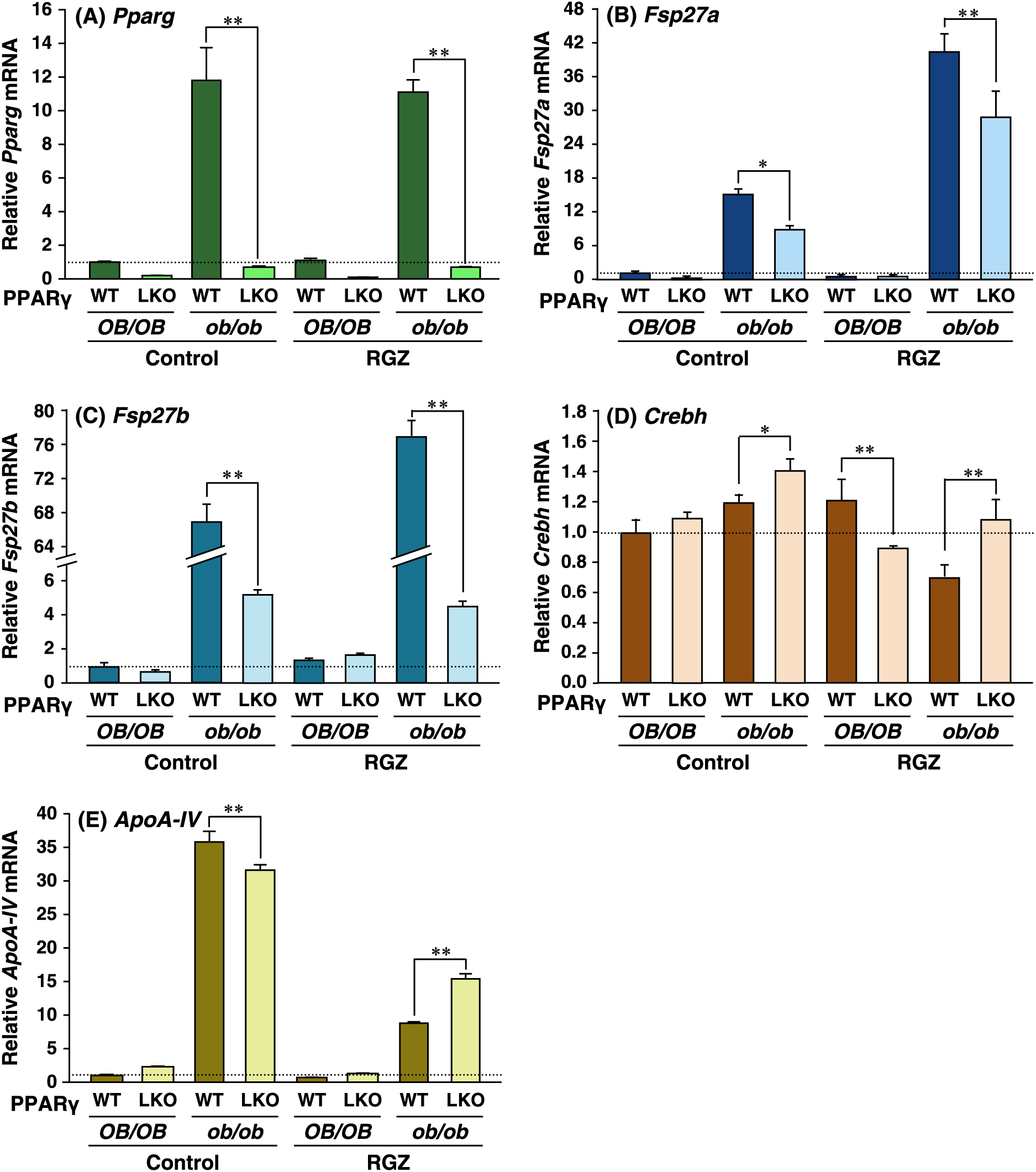
The fasting condition induced not only Fsp27b but also Fsp27a, and refeeding led to a marked decrease in the expression of both genes (Fig. 2A and B). To elucidate whether hepatic PPARγ is associated with the fasting induction of the expression of both Fsp27b and Fsp27a, OB/OB-PPARγLKO mice were fasted. Pparg mRNA expression was decreased in the fasting OB/OB-PPARγWT liver and was at a basal level in OB/OB-PPARγLKO mice (Fig. 2C). Expression of Fsp27a and Fsp27b mRNAs in OB/OB-PPARγWT mice was induced to approximately 2- and 20-fold in fasting mice compared to that in non-fasting (control) mice (Fig. 2D and E). However, both Fsp27a and Fsp27b mRNAs in OB/OB-PPARγLKO mice showed a tendency for higher expression levels as compared to those in OB/OB-PPARγWT mice, indicating the involvement of another factor besides PPARγ in the induction of both genes under fasting conditions.
Effect of forced expression of PPARγ on Fsp27a and Fsp27b expression in AML-12 cells
To gain more evidence for Fsp27b regulation by PPARγ, an adenovirus expressing PPARγ (AdPPARγ) and control LacZ (AdLac) was used to infect the liver-derived cell line AML-12, which does not constitutively express Fsp27a/b or Pparg. Ap2 and Fsp27a genes are typical PPARγ-target genes, which are abundantly expressed in adipocytes [11]. Although the expression of these genes was not observed in AdLac-infected cells, both genes were elevated in AdPPARγ-infected cells without RGZ and markedly induced in AdPPARγ-infected cells with RGZ (Fig. 3A and B). Fsp27b also showed the same tendency and was induced by approximately 20-fold in AdPPARγ-infected cells with or without RGZ (Fig. 3C). Crebh mRNA was decreased in AdPPARγ-infected cells without RGZ and increased in AdPPARγ-infected cells with RGZ (Fig. 3D). These results suggest that the expression of hepatic PPARγ not only causes an increase in the expression of Fsp27a mRNA, but also of Fsp27b mRNA in AML-12 cells.

Previously, we identified a functional PPRE (–214/–202) located on the Fsp27a promoter [11]. In addition, CREBH response element (CHRE, –35/–27) located on the Fsp27b promoter (Fig. 4A) [14]. The PPRE position was revealed at –1,163/–1,151 from the transcriptional start site (+1) of Fsp27b (Fig. 4A). However, the existence of a functional PPRE on the Fsp27b 5'-flanking region remains unclear. To determine how PPARγ regulates the promoter activity of Fsp27b, a Fsp27b D1 (–2.7 kb/+1 bp) luciferase reporter was firstly constructed. The Fsp27b D1 construct and PPARγ and RXRα expression plasmids were transfected into liver-derived AML-12 cells. The fold-inductions of Fsp27b promoter activity in AML-12 cells with PPARγ, PPARγ + RXRα, and PPARγ + RXRα + RGZ compared to those in AML-12 cells without PPARγ were 1.5-fold, 1.9-fold, and 4.2-fold, respectively (Fig. 4Ba).
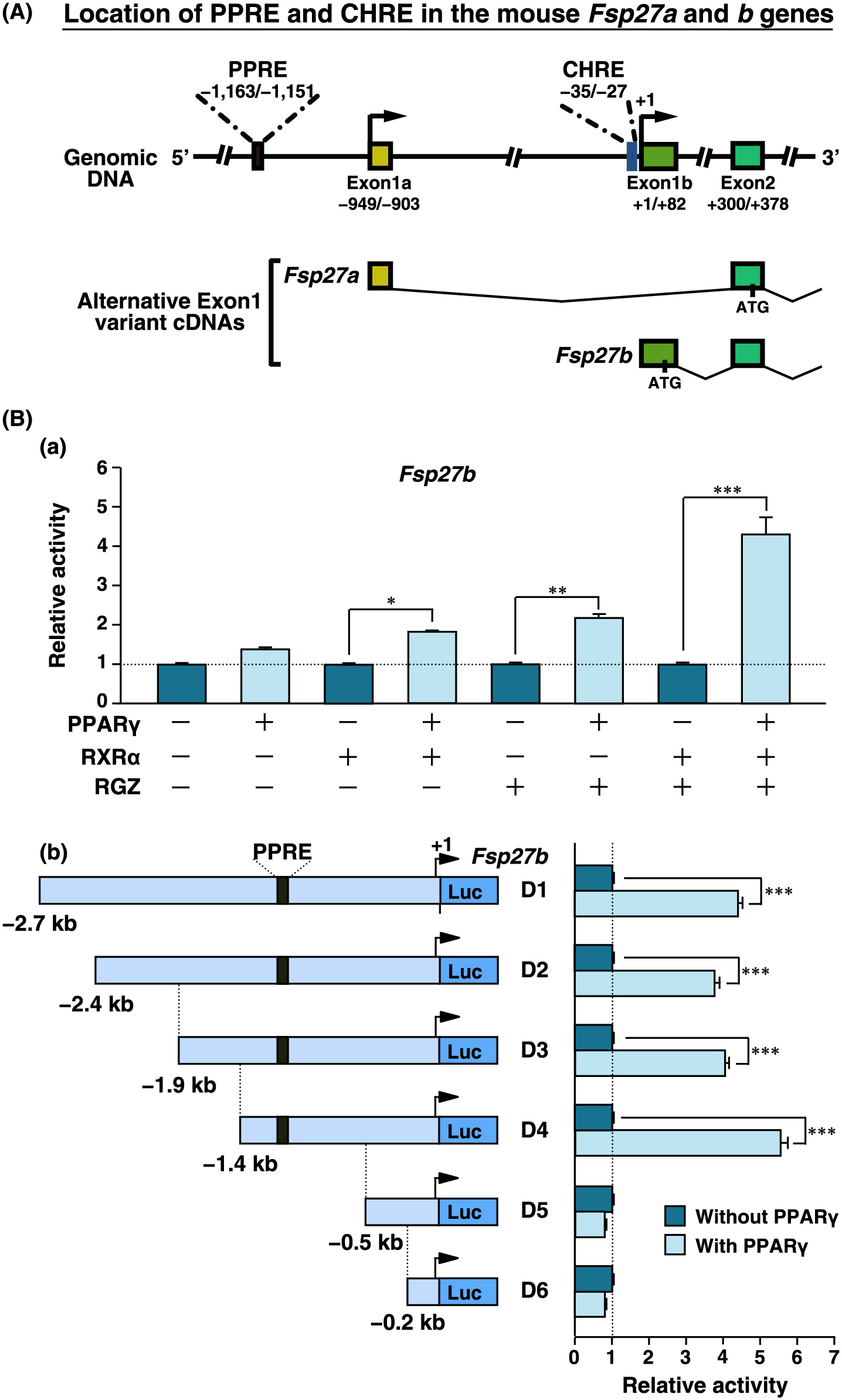
Furthermore, to identify the cis-element responsible for PPARγ-dependent promotion of Fsp27b expression, reporter constructs with serial deletions of the 5'-flanking regions of Fsp27b were prepared. The luciferase activities of Fsp27b D1–D4 constructs were induced by approximately 4–5.5-fold with PPARγ compared to those without PPARγ, whereas the induction of the D5 and D6 constructs lacking PPRE was completely lost (Fig. 4Bb). These results suggest that PPARγ induces the promoter activity of Fsp27b through PPRE, which is present on the Fsp27a promoter.
Discussion
Recently, FSP27 isoforms were identified as FSP27α and FSP27β. Fsp27a and Fsp27b have a different promotor region [14], suggesting that transcriptional regulation of Fsp27b differs from that of Fsp27a. We previously demonstrated that PPARγ directly regulates Fsp27a expression in fatty liver of ob/ob mice and determined the functional PPRE (–214/–202 bp) on the promotor region, which upregulates PPARγ-dependent promoter activity and directly binds to PPARγ [11]. Upon analysis of the genome sequences of Fsp27a and Fsp27b, the PPRE of Fsp27a corresponded to the –1,163/–1,151 bp of the Fsp27b 5'-flanking region. However, it remains unclear whether Fsp27b in fatty liver is regulated by PPARγ or whether the PPRE located on the Fsp27b 5'-flanking region is also functional for the promotor activity. The present study demonstrated that PPARγ regulates Fsp27b in fatty liver of ob/ob mice in vivo, but not in fasting-inducible liver. Furthermore, Fsp27b regulation by PPARγ was promoted through the PPRE located in the Fsp27b 5'-flanking region. These results suggest that hepatic PPARγ regulates the expression of not only Fsp27a but also of Fsp27b through a common PPRE in the fatty liver of ob/ob mice.
In contrast to the potential function of PPARγ in Fsp27a/b expression in the fatty liver of ob/ob mice, PPARγ in the liver of fasting mice is likely to not be involved in the induction of Fsp27a/b because the expression of the latter was markedly induced by fasting under PPARγ deficiency. However, the mechanism involved in the induction of Fsp27a/b mRNA by the fasting condition in liver lacking PPARγ remains unclear.
An earlier study on CREBH-null mice clearly demonstrated that CREBH plays an essential role in the induction of Fsp27b in the liver of fasting mice. It is well known that hepatic Crebh mRNA expression is markedly induced under fasting, and suppressed after refeeding [16-18]. Fsp27b was also expressed at a high level in fatty liver of ob/ob mice [14], although the contribution of CREBH toward Fsp27b expression in the fatty liver of ob/ob mice remains unclear. Our results suggested that hepatic PPARγ and CREBH independently upregulate Fsp27b expression in the fatty liver of ob/ob mice. It is possible that PPARγ regulates CREBH expression because a functional PPRE is located on the CREBH promoter [16, 18]. However, it is not likely that CREBH expression in the fatty liver of ob/ob mice is regulated by PPARγ since the Crebh mRNA remained unchanged in the ob/ob-PPARγLKO liver. Interestingly, an earlier report also revealed that Fsp27a was not detected in fatty liver of ob/ob mice [14]. In the present study, the expression of Fsp27a was induced in fatty liver of ob/ob mice as compared with that in control lean mice, although the relative fold-induction of Fsp27a was lower than that of Fsp27b. The reason for the discrepancy between our results and those of this previous study remains unclear. The age of the ob/ob mice used in each study may be one of the factors affecting hepatic Fsp27a expression.
A limitation of this study is that we could not examine FSP27α/β protein levels in all liver samples. We were unable to detect endogenous FSP27α/β protein in the liver because not all commercial FSP27α/β IgGs were suitable for western blot analysis owing to the presence of high background or many extra bands under the present conditions. The detection or estimation of constitutive FSP27 protein in liver and cell samples will be the subject of future investigation.
In summary, the present study demonstrated that hepatic Fsp27a and Fsp27b are positively regulated by PPARγ through a common functional PPRE. Although the expression of Fsp27a/b in the liver under normal conditions is maintained at low levels, their expression in fatty liver is markedly increased through the elevation of hepatic PPARγ expression. Therefore, hepatic PPARγ-regulated Fsp27a/b is likely to largely contribute to the aggravation of diabetic fatty liver. Moreover, CREBH appears to be an important factor affecting fasting-inducible Fsp27b expression. Since CIDEC, the human homolog of mouse FSP27, also increases in fatty liver [19], elucidating the mechanism of TG-accumulation effects on FSP27α/β may lead to new therapeutic opportunities for controlling TG accumulation in non-alcoholic fatty liver disease and its associated pathologies.
Acknowledgments
This work was supported by a Grant from KAKENHI (17K08799). We would like to thank Editage (www.editage.jp) for English language editing.
Disclosure
None of the authors have any potential conflicts of interest associated with this research.
References
- 1 Danesch U, Hoeck W, Ringold GM (1992) Cloning and transcriptional regulation of a novel adipocyte-specific gene, FSP27. CAAT-enhancer-binding protein (C/EBP) and C/EBP-like proteins interact with sequences required for differentiation-dependent expression. J Biol Chem 267: 7185–7193.
- 2 Williams PM, Chang DJ, Danesch U, Ringold GM, Heller RA (1992) CCAAT/enhancer binding protein expression is rapidly extinguished in TA1 adipocyte cells treated with tumor necrosis factor. Mol Endocrinol 6: 1135–1141.
- 3 Liang L, Zhao M, Xu Z, Yokoyama KK, Li T (2003) Molecular cloning and characterization of CIDE-3, a novel member of the cell-death-inducing DNA-fragmentation-factor (DFF45)-like effector family. Biochem J 370: 195–203.
- 4 Puri V, Konda S, Ranjit S, Aouadi M, Chawla A, et al. (2007) Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J Biol Chem 282: 34213–34218.
- 5 Jambunathan S, Yin J, Khan W, Tamori Y, Puri V (2011) FSP27 promotes lipid droplet clustering and then fusion to regulate triglyceride accumulation. PLoS One 6: e28614.
- 6 Sun Z, Gong J, Wu H, Xu W, Wu L, et al. (2013) Perilipin1 promotes unilocular lipid droplet formation through the activation of Fsp27 in adipocytes. Nat Commun 4: 1594.
- 7 Nishino N, Tamori Y, Tateya S, Kawaguchi T, Shibakusa T, et al. (2008) FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J Clin Invest 118: 2808–2821.
- 8 Toh SY, Gong J, Du G, Li JZ, Yang S, et al. (2008) Up-regulation of mitochondrial activity and acquirement of brown adipose tissue-like property in the white adipose tissue of fsp27 deficient mice. PLoS One 3: e2890.
- 9 Tanaka N, Takahashi S, Matsubara T, Jiang C, Sakamoto W, et al. (2015) Adipocyte-specific disruption of fat-specific protein 27 causes hepatosteatosis and insulin resistance in high-fat diet-fed mice. J Biol Chem 290: 3092–3105.
- 10 Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, et al. (2003) Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest 111: 737–747.
- 11 Matsusue K, Kusakabe T, Noguchi T, Takiguchi S, Suzuki T, et al. (2008) Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metab 7: 302–311.
- 12 Vila-Brau A, De Sousa-Coelho AL, Goncalves JF, Haro D, Marrero PF (2013) Fsp27/CIDEC is a CREB target gene induced during early fasting in liver and regulated by FA oxidation rate. J Lipid Res 54: 592–601.
- 13 Xu MJ, Cai Y, Wang H, Altamirano J, Chang B, et al. (2015) Fat-specific protein 27/CIDEC promotes development of alcoholic steatohepatitis in mice and humans. Gastroenterology 149: 1030.e6–1041.e6.
- 14 Xu X, Park JG, So JS, Lee AH (2015) Transcriptional activation of Fsp27 by the liver-enriched transcription factor CREBH promotes lipid droplet growth and hepatic steatosis. Hepatology 61: 857–869.
- 15 Xu X, Park JG, So JS, Hur KY, Lee AH (2014) Transcriptional regulation of apolipoprotein A-IV by the transcription factor CREBH. J Lipid Res 55: 850–859.
- 16 Danno H, Ishii KA, Nakagawa Y, Mikami M, Yamamoto T, et al. (2010) The liver-enriched transcription factor CREBH is nutritionally regulated and activated by fatty acids and PPARalpha. Biochem Biophys Res Commun 391: 1222–1227.
- 17 Lee MW, Chanda D, Yang J, Oh H, Kim SS, et al. (2010) Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab 11: 331–339.
- 18 Kim H, Mendez R, Zheng Z, Chang L, Cai J, et al. (2014) Liver-enriched transcription factor CREBH interacts with peroxisome proliferator-activated receptor α to regulate metabolic hormone FGF21. Endocrinology 155: 769–782.
- 19 Hall AM, Brunt EM, Klein S, Finck BN (2010) Hepatic expression of cell death-inducing DFFA-like effector C in obese subjects is reduced by marked weight loss. Obesity (Silver Spring) 18: 417–419.

