Abstract
Muscle wasting is a complication in patients with diabetes and leads to a reduced quality of life. However, the detailed mechanisms of diabetes-induced muscle wasting remain unknown. Plasminogen activator inhibitor-1 (PAI-1), a serine protease inhibitor that suppresses plasminogen activator activity, is involved in the pathophysiology of various diseases, including diabetes. In the present study, we examined the role of endogenous PAI-1 in the decrease in muscle mass and the impaired grip strength induced by the diabetic state by employing streptozotocin (STZ)-treated PAI-1-deficient female mice. The analyses of skeletal muscles and grip strength were performed in PAI-1-deficient and wild-type mice 4 weeks after the induction of a diabetic state by STZ administration. PAI-1 deficiency did not affect muscle mass in the lower limbs measured by quantitative computed tomography or tissue weights of the tibialis anterior, gastrocnemius and soleus muscles of female mice with or without STZ treatment. On the other hand, PAI-1 deficiency significantly aggravated grip strength decreased by STZ in female mice. PAI-1 deficiency did not affect the mRNA levels of Pax7, MyoD, myogenin or myosin heavy chain in either the tibialis anterior or soleus muscles of female mice with or without STZ treatment. In conclusion, we revealed for the first time that PAI-1 deficiency aggravates grip strength impaired by the diabetic state in female mice, although it did not affect diabetes-decreased muscle mass.
DIABETES induces various complications, such as nephropathy, retinopathy, neuropathy, peripheral angiopathy and cardiovascular diseases [1]. In addition to these complications, the diabetic state is related to muscle wasting [2-5]. Diabetic muscle wasting can increase the risk of falls and fractures, which may lead to a reduced quality of life and an increased risk of death. Morphologically asymptomatic myofiber atrophy was observed in type 1 diabetic patients with neuropathy [6], suggesting that diabetic conditions affect skeletal muscle through metabolic changes prior to neuropathy complications. Muscle mass is regulated by muscle differentiation and muscle protein amount. It has been suggested that diabetic sarcopenia is caused by impaired muscle differentiation and an imbalance between protein synthesis and degradation caused by metabolic changes, such as hyperglycemia, decreased insulin action and increased advanced glycation end products [5]. However, the detailed mechanisms of diabetes-induced muscle wasting are not fully understood.
Plasminogen activator inhibitor-1 (PAI-1) is a serine protease inhibitor that suppresses plasminogen activator activity and inhibits fibrinolysis [7]. PAI-1 is also known to be involved in several metabolic disorders, such as cardiovascular diseases, diabetes and osteoporosis, as an adipokine [7]. PAI-1 has been reported to participate in the response to skeletal muscle injury and myopathic conditions [8-10]. Krause et al. reported that PAI-039, a pharmacological PAI-1 inhibitor, improves muscle repair after muscle injury impaired by diabetes in mice [9]. We previously revealed in PAI-1-deficient mice that PAI-1 is involved in osteopenia and delayed bone repair induced by diabetes in female mice [11-13]. Moreover, we showed that PAI-1 is involved in glucose metabolism abnormalities, muscle wasting, osteopenia and delayed bone repair induced by glucocorticoid excess in mice [14-16]. Taken together, PAI-1 might participate in muscle wasting induced by diabetes. However, there have been no reports available about the relationships between diabetic muscle wasting and PAI-1.
In this study, therefore, we investigated the roles of PAI-1 in muscle wasting induced by diabetes using streptozotocin (STZ)-treated PAI-1-deficient female mice.
Materials and Methods
Materials
STZ was purchased from Sigma-Aldrich Japan (Tokyo, Japan). All other chemicals used were of analytical grade.
Animal experiment
Eight-week-old female or male PAI-1-deficient (KO) mice and their wild-type counterparts [C57BL/6J (81.25%) and 129/SvJ (18.75%) background] were used. These mice were provided by Professor D. Collen (University of Leuven, Leuven, Belgium). Diabetes was induced four times in 8-week-old mice by intraperitoneal injection of the pancreatic β-cell toxin STZ (200 mg/kg body weight in saline) twice weekly [13]. Controls were injected with saline alone. Four days after the last injection (day 4), the nonfasting blood glucose level was measured with a glucometer (Glutest Ace, Sanwa Kagaku Kenkyusyo, Nagoya, Japan) by using blood obtained from the tail vein. Mice with blood glucose levels greater than 300 mg/dL were considered diabetic. Blood glucose levels were confirmed at least twice. Animals were maintained in metabolic cages on a 12-h light, 12-h dark cycle, and they received food and water ad libitum. At 4 weeks after the induction of diabetes, quantitative computed tomography (qCT) analysis was performed to measure lower limb muscle mass, and all mouse (placebo- and STZ-treated groups) muscle tissues were obtained. All animal experiments were performed according to the guidelines of the National Institute of Health (NIH) and the institutional rules for the use and care of laboratory animals of Kindai University.
In vivo quantitative computed tomography (qCT) analysis
Mice were scanned using a LaTheta (LCT-200) experimental animal CT system (Hitachi-Aloka Medical, Tokyo, Japan), as previously described [17]. Briefly, regions of interest (ROIs) were defined as the whole body for the assessment of the muscle mass around the tibia. The parameters used for the CT scans were as follows: tube voltage, 50 kVp; tube current, 500 μA; integration time, 3.6 ms; axial field of view, 48 mm; voxel size, 96 × 192 × 1,008 μm. Muscle mass was analyzed using LaTheta software (version 3.40).
Histological analysis
After euthanization, the muscles were collected from the dead mice, fixed in 4% paraformaldehyde in phosphate buffer (pH 7.4) for 16 h at 4°C, and further fixed for 7 days in 80% ethanol. The muscles were embedded in paraffin. Four-micrometer-thick sections were obtained and stained with hematoxylin/eosin. The hematoxylin/eosin-stained sections were photographed with a microscope, and cross-sectional areas of at 250 (soleus muscle) or 500 (tibialis anterior muscle) myofibers were quantified by using ImageJ.
Measurement of grip strength
We measured grip strength with a pull bar attached to a grip strength meter (1-27SM, Columbus Instruments, Columbus, OH, USA), as described previously [18]. A pull bar attachment was grasped by the four limbs of a mouse and was then pulled at approximately 2 cm/sec. The test was repeated 5 times, and the data represented the average of each mouse.
Real‑time polymerase chain reaction (PCR) analysis
Total RNA was extracted from homogenized samples of the muscle tissues (and the cultured cells) using an RNeasy mini kit for fibrous tissue (Life Technologies Japan, Tokyo, Japan). Real-time PCR was performed using StepOne Plus and Fast SYBR Green PCR Master mix (Life Technologies Japan, Tokyo, Japan) as previously described [13]. The following primer sets were used: Pax7, forward: 5'-CCCTCAGTGAGTTCGATTAGCC-3', reverse: 5'-GGTCGGGTTCTGATTCCACA-3'; MyoD, forward: 5'-AGCACTACAGTGGCGACTCAG-3'; reverse: 5'-AGGCGGTGTCGTAGCCATTC-3'; myogenin, forward: 5'-GCTGCCTAAAGTGGAGATCCT-3', reverse: 5'-GCGCTGTGGGAGTTGCAT-3'; myosin heavy chain 1 (MHC-1), forward: 5'-CTCTTCCCGCTTTGGTAAGTT-3', reverse: 5'-CAGGAGCATTTCGATTAGATCCG-3'; myosin heavy chain 2b (MHC-2b), atrogin1, forward: 5'-GCAGAGAGTCGGCAAGTC-3', reverse: 5'-CAGGTCGGTGATCGTGAG-3'; insulin-like growth factor-1 (IGF-1), forward: 5'-CAAGCCCACAGGCTATGGC-3', reverse: 5'-TCTGAGTCTTGGGCATGTCAG-3'; fibroblast growth factor2 (FGF2), forward: 5'-GCGACCCACACGTCAAACTA-3', reverse: 5'-CCGTCCATCTTCCTTCATAGC-3'; FNDC5, forward: 5'-TCATTGTTGTGGTCCTCTTC-3', reverse: 5'-GCTCGTTGTCCTTGATGATA-3'; PAI-1, forward: 5'-TTCAGCCCTTGCTTGCCTC-3', reverse: 5'-ACACTTTTACTCCGAAGTCGGT-3'; β-actin, forward: 5'-TACCACAGGCATTGTGATGG-3', reverse: 5'-TTTGATGTCACGCACGATTT-3'. The mRNA levels in the tissues of mice were normalized relative to the amount of β-actin.
Statistical analysis
Data are represented as the means ± standard errors of the mean (SEM). Statistical analyses were performed by the Mann-Whitney U test for comparisons of 2 groups. Two-way ANOVA followed by Tukey’s test was performed for multiple comparisons. Differences of p < 0.05 were regarded as significant. All statistical analyses were performed using StatView version 5.0 software (Statistical Analysis System (SAS) Institute Inc.; Cary, NC, USA).
Results
Effect of PAI-1 deficiency on muscle mass decreased by diabetic state
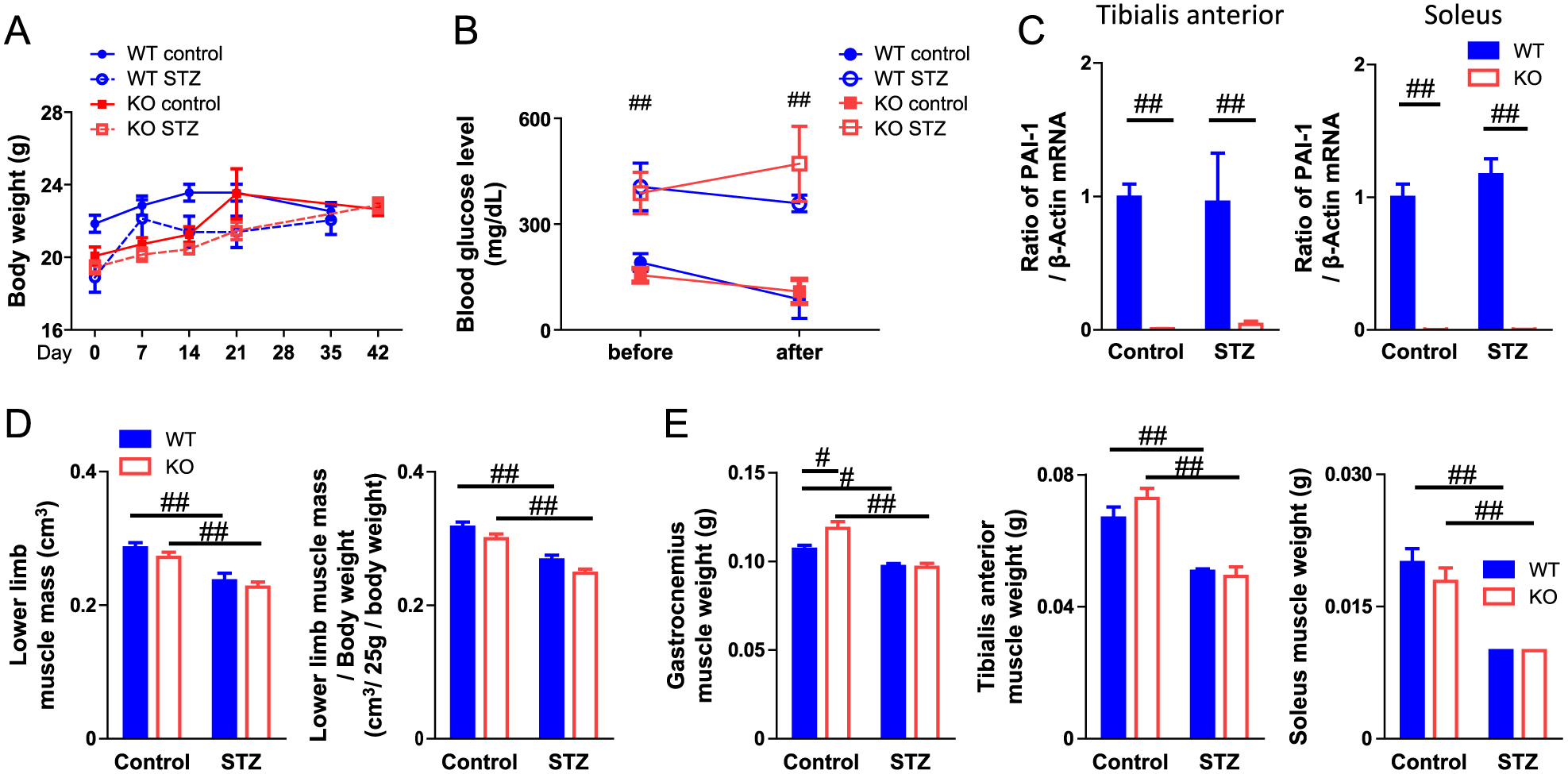
PAI-1 deficiency did not affect body weight or blood glucose levels with or without STZ administration in female mice (Fig. 1A, B). STZ did not affect PAI-1 mRNA levels in the tibialis anterior and soleus muscles, although PAI-1 expression was not detected in PAI-1 KO mice (Fig. 1C). We examined the effects of PAI-1 deficiency on muscle mass in mice with or without diabetes. As shown in Fig. 1D, PAI-1 deficiency did not affect muscle mass or body weight-adjusted muscle mass in the lower limbs of mice with or without STZ administration, as measured by qCT (Fig. 1D). Moreover, PAI-1 deficiency did not affect the tissue weights of the tibialis anterior, gastrocnemius and soleus muscles of mice, which were decreased by the diabetic state (Fig. 1E). PAI-1 deficiency did not affect the appearance and cross-sectional areas of muscle fibers in either the tibialis anterior or soleus muscles of mice with or without diabetes (Fig. 2A, B).

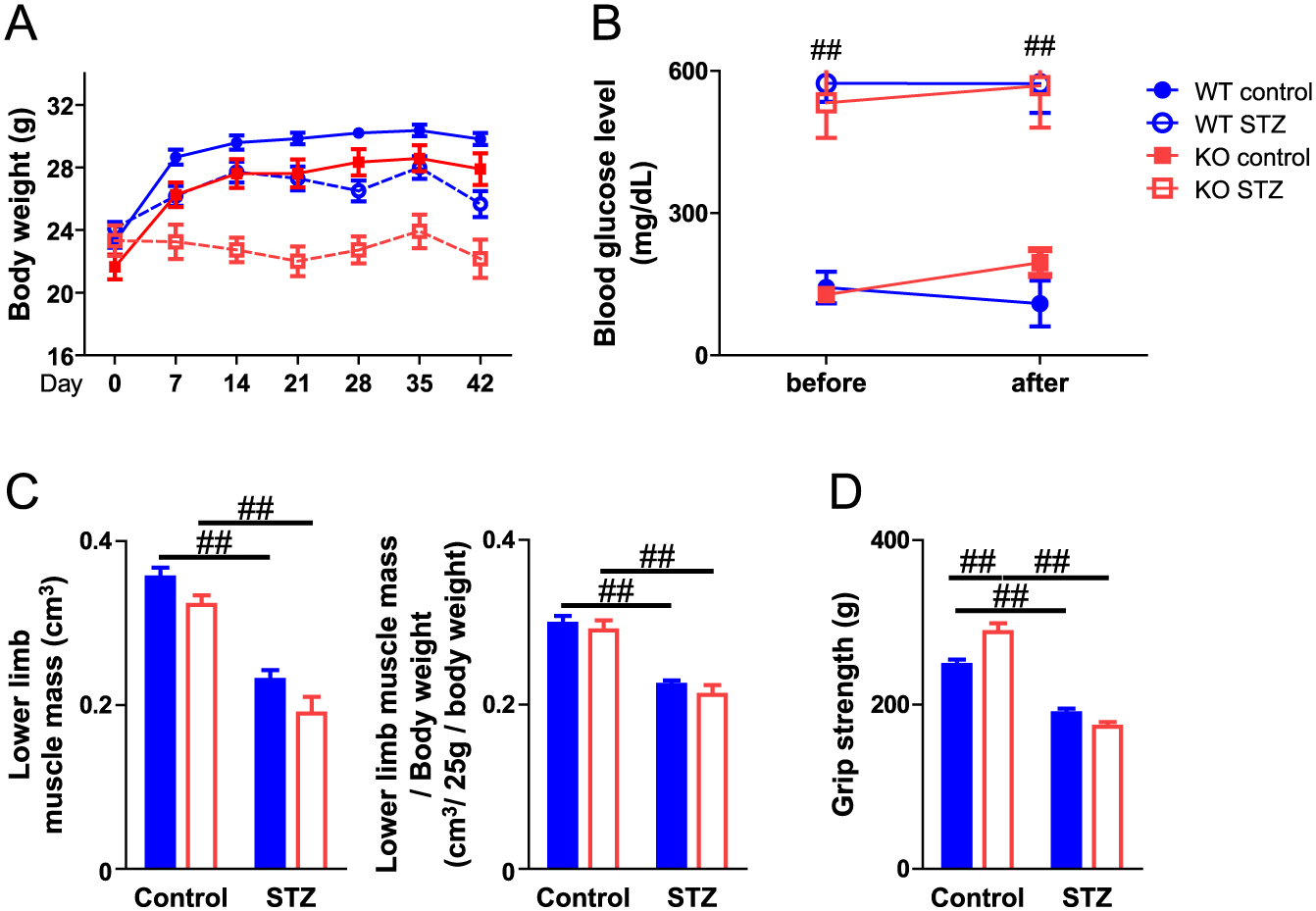
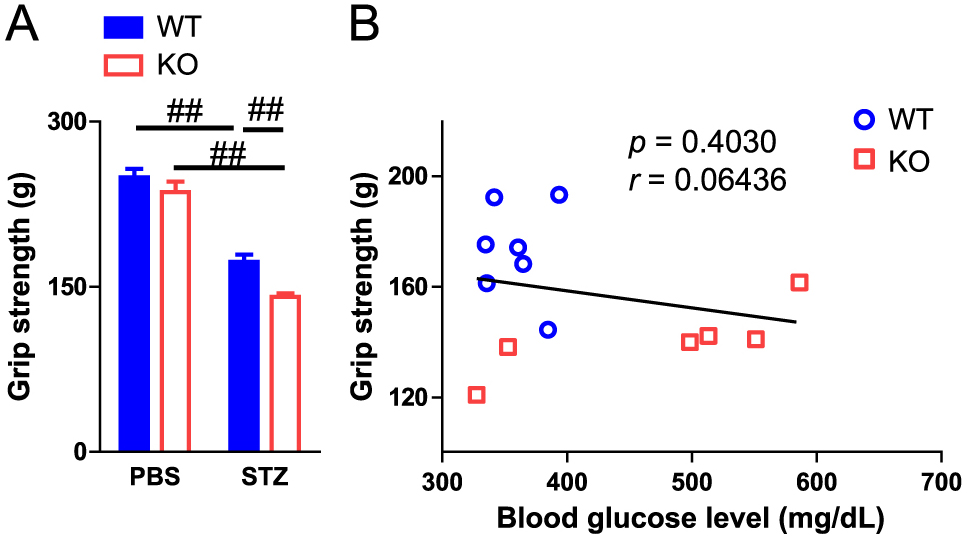
Sarcopenia and muscle wasting induced by the pathological state are influenced by a decrease in muscle mass and function. Muscle strength is one of the most important components of muscle function. We next examined the effects of PAI-1 deficiency on limb grip strength to evaluate the role of PAI-1 in muscle strength in female mice. As shown in Fig. 3A, STZ administration significantly decreased the grip strength of mice. PAI-1 deficiency significantly aggravated grip strength decrease in the diabetic state in female mice (Fig. 3A). Since blood glucose levels might influence grip strength, we evaluated the relationships between blood glucose levels and grip strength of female mice with STZ treatment using simple regression analysis. However, grip strength did not correlate with blood glucose levels in mice administered STZ (Fig. 3B). Obvious hemorrhage as well as changes in appearance, size, behavior and food intake were not observed. On the other hand, PAI-1 deficiency did not affect the decrease in muscle mass in the lower limbs or the decrease in grip strength in male mice with or without diabetes (Fig. 4).
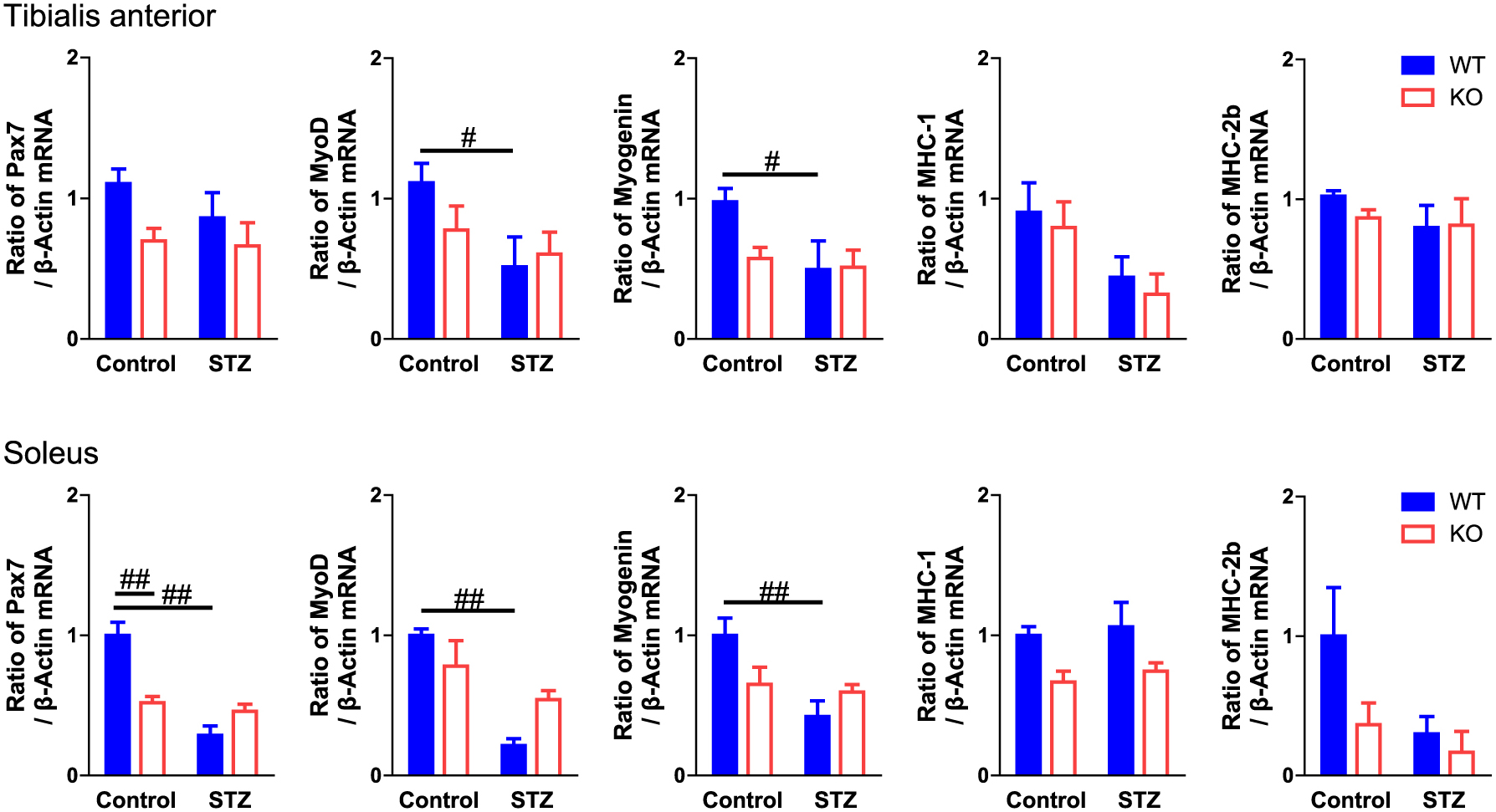
Muscle differentiation regulates muscle mass in physiological and pathophysiological states. Pax7 is a muscle satellite cell-specific marker, and muscle differentiation is accelerated by myogenic regulatory factors, such as MyoD and myogenin [19]. Myotubes express MHC abundantly [20]. We examined the effects of PAI-1 deficiency on the expression of Pax7, MyoD, myogenin, MHC-1 and MHC-2b in the tibialis anterior and soleus muscles of female mice with or without STZ treatment. STZ significantly suppressed the mRNA levels of MyoD and myogenin in both the tibialis anterior and soleus muscles of mice, although STZ did not affect the mRNA levels of MHC-1 and MHC-2b in either muscle (Fig. 5). In addition, STZ significantly suppressed Pax7 mRNA levels in the soleus muscles of mice but not in the tibialis anterior. PAI-1 deficiency did not affect the mRNA levels of Pax7, MyoD, myogenin, MHC-1 or MHC-2b in either the tibialis anterior or soleus muscles of mice with or without diabetes (Fig. 5).
Our previous study suggested that the diabetic state might affect the expression of some myokines, such as irisin, IGF-1 and FGF2, in female mice [21]. We therefore examined the effects of PAI-1 deficiency on the expression of irisin, a proteolytic product of FNDC5, as well as IGF-1 and FGF2 in the tibialis anterior and soleus muscles of female mice with or without STZ treatment. As shown in Fig. 6, PAI-1 deficiency significantly reduced FNDC5 mRNA levels, although it did not affect FNDC5 mRNA levels suppressed by the diabetic state in either the tibialis anterior or soleus muscles of mice. PAI-1 deficiency significantly enhanced IGF-1 mRNA levels in the diabetic state only in the tibialis anterior muscles but not the soleus muscles. Moreover, PAI-1 deficiency significantly enhanced FGF-2 mRNA levels with or without STZ treatment in the tibialis anterior muscles of mice, although it significantly reduced FGF2 mRNA levels in the soleus muscles in the state without STZ treatment (Fig. 6).

Discussion
Sarcopenia is characterized by decreased muscle mass and impaired muscle strength and function [22]. We previously revealed that PAI-1 deficiency blunts muscle mass and that MyoD expression is suppressed by glucocorticoid excess in the type IIb muscle fiber-dominant gastrocnemius of mice but not in the type I muscle fiber-dominant soleus muscles [14, 15]. Since excess GC induces glucose metabolism abnormalities, we speculated that PAI-1 might be involved in decreased muscle mass induced by the diabetic state. However, we showed that PAI-1 deficiency does not affect the lower limb muscle mass or tissue weight of type IIb muscle fiber-dominant tibialis anterior/gastrocnemius muscles and type 1 muscle fiber-dominant soleus muscles decreased by STZ treatment in female mice in the present study. Moreover, PAI-1 deficiency did not affect the expression of MyoD, and myogenin decreased by STZ treatment in both the tibialis anterior and soleus muscles of female mice. These findings indicate that PAI-1 is not involved in the decrease in muscle mass induced by the diabetic state in female mice. Although PAI-1 is involved in various diseases as an adipokine produced by adipose tissues [7], it is well known that STZ-treated mice possess less adipose tissue than glucocorticoid-treated or obese mice. Moreover, our previous study suggested that adipose tissue-derived PAI-1 might be related to glucocorticoid-induced muscle wasting and bone loss in mice [14]. We therefore cannot rule out the possibility that PAI-1 might be related to decreased muscle mass induced by the diabetic state in obese mice. In our data, PAI-1 deficiency significantly decreased Pax7 expression in the soleus muscles of PAI-1-untreated control mice, and this effect was not observed in STZ-treated mice. This finding is not compatible with previous evidence showing that pharmacological inhibition of PAI-1 restores muscle regeneration after injury in mice, although gene-deleted mice were not used in that study [9]. Despite the fact that the reason for this discrepancy remains unknown and is not the topic of our study, further studies using PAI-1-deficient mice will be necessary to clarify the role of PAI-1 in muscle regeneration after injury.
Grip strength is the most useful measurement of muscle strength for the evaluation of sarcopenia in humans and mice. The present study showed that PAI-1 deficiency significantly aggravates the decrease in grip strength induced by STZ treatment in female mice. Previous studies indicate that skeletal muscles produce PAI-1 locally [8, 22, 23]. Since the relationships between blood glucose level and grip strength in STZ-treated mice were not significant in the simple regression analysis, we speculated that the effects of PAI-1 deficiency on decreased grip strength are not due to an elevation in blood glucose levels. The physiological roles of endogenous PAI-1 in muscle strength remain unknown in our study. Although circulating PAI-1 elevated by pathological states, such as diabetes, glucocorticoid excess and inflammation, negatively affects various tissues, including the musculoskeletal system, there has been previous evidence about the protective roles of endogenous PAI-1 in the musculoskeletal system in physiological and pathological states [24-27]. PAI-1 transgenic mice have increased bone size, and pharmacological PAI-1 inhibition suppresses the differentiation of mesenchymal stem cells into chondrocytes in vitro [24, 25]. Moreover, we revealed that PAI-1 is involved in the differentiation of mesenchymal stem cells into osteoblasts in an in vitro study using cells from PAI-1-deficient mice [27]. In addition, we reported that PAI-1 deficiency enhances subchondral osteopenia after the induction of osteoarthritis in mice, suggesting that PAI-1 might play a protective role in the subchondral bone in knee osteoarthritis [26]. We speculated that endogenous PAI-1 fulfills a protective role in diabetes-induced disturbance of muscle function in female mice.
It has been recently noted that myokines produced from skeletal muscles play some physiological and pathophysiological roles in the skeletal system [28, 29]. We previously showed that a diabetic state decreases the expression of IGF-1, FGF-2 and irisin in the gastrocnemius muscles of mice [21]. In the present study, PAI-1 deficiency enhanced IGF-I expression in the tibialis anterior muscles of female mice treated with STZ. Moreover, PAI-1 deficiency significantly enhanced FGF-2 expression in the tibialis anterior muscles of female mice with or without STZ treatment. Considering that PAI-1 deficiency enhances a decrease in muscle strength but not a decrease in muscle mass induced by the diabetic state in female mice, the regulation of muscle-derived factors, such as IGF-1 and FGF-2, might partly contribute to the inhibition of some protective effects of endogenous PAI-1 on muscle mass. For irisin expression, we showed that PAI-1 deficiency significantly suppresses FNDC5 expression. Moreover, STZ treatment suppressed FNDC5 expression with or without PAI-1 deficiency in both the tibialis anterior and soleus muscles of female mice. These findings suggest that endogenous PAI-1 is partly related to irisin production in the muscles of female mice, although its physiological significance is unknown. Since the diabetic state did not affect PAI-1 expression in muscles in our study, it is not likely that the diabetic state suppresses the effects of endogenous PAI-1 on irisin expression. The diabetic state might suppress irisin production independently of PAI-1 in mice.
Sex differences exist in muscle size, muscle development and muscle mass. There is evidence suggesting sex differences in bone metabolism. A previous study suggested that differences in osteoblast phenotypes between male and female osteoblasts might exist at the genetic level, but not at sex hormones [30]. We recently identified Serpina3n, which has inhibitory activity on the phenotypes of differentiated osteoblasts as the most female osteoblast-dominant gene in mice [31]. As for PAI-1, we previously reported that PAI-1 is involved in diabetes-induced osteoporosis in female mice, but not in male mice [11]. In that study, PAI-1 inhibited osteoblast differentiation, alkaline phosphatase activity and mineralization in mouse primary osteoblasts from female mice, but not in those from male mice. In the present study, PAI-1 deficiency aggravated grip strength decreased by STZ in female mice, but not in male mice, which was similar with the effects of PAI-1 on bone in mouse. Alternatively, the effects of PAI-1 deficiency itself might antagonize their augmentation of the decrease in grip strength induced by diabetes in male mice, because PAI-1 deficiency slightly increased grip strength in male mice, but not in female mice, in our data. Further study is necessary to clarify the mechanism of sex differences for the role of PAI-1 in sarcopenia.
PAI-1 plays various physiological and pathophysiological roles in numerous diseases, including diabetes [7]. Therefore, the effects of PAI-1 deficiency on muscle in our study might be due to factors other than PAI-1, although PAI-1 deficiency did not affect the body weight, blood glucose level, hemorrhage, appearance, size, behavior or food intake of mice with or without diabetes.
In conclusion, we showed that PAI-1 deficiency aggravates grip strength impaired by the diabetic state in female mice, although it did not affect diabetes-decreased muscle mass. PAI-1 might protect against diabetes-induced muscle dysfunction in mice.
Acknowledgments
This study was supported in part by a Grant-in-Aid for Scientific Research (C:20K09514) to H.K. from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We acknowledge Katsumi Okumoto (Life Science Research Institute, Kindai University) for his technical assistance.
Disclosure Statement
The authors declare no conflicts of interest.
References
- 1 Ahlqvist E, van Zuydam NR, Groop LC, McCarthy MI (2015) The genetics of diabetic complications. Nat Rev Nephrol 11: 277–287.
- 2 Park SW, Goodpaster BH, Strotmeyer ES, de Rekeneire N, Harris TB, et al. (2006) Decreased muscle strength and quality in older adults with type 2 diabetes: the health, aging, and body composition study. Diabetes 55: 1813–1818.
- 3 Park SW, Goodpaster BH, Strotmeyer ES, Kuller LH, Broudeau R, et al. (2007) Accelerated loss of skeletal muscle strength in older adults with type 2 diabetes: the health, aging, and body composition study. Diabetes Care 30: 1507–1512.
- 4 Park SW, Goodpaster BH, Lee JS, Kuller LH, Boudreau R, et al. (2009) Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 32: 1993–1997.
- 5 Krause MP, Riddell MC, Hawke TJ (2011) Effects of type 1 diabetes mellitus on skeletal muscle: clinical observations and physiological mechanisms. Pediatr Diabetes 12: 345–364.
- 6 Reske-Nielsen E, Harmsen A, Vorre P (1977) Ultrastructure of muscle biopsies in recent, short-term and long-term juvenile diabetes. Acta Neurol Scand 55: 345–362.
- 7 Kaji H (2016) Adipose tissue-derived plasminogen activator inhibitor-1 function and regulation. Compr Physiol 6: 1873–1896.
- 8 Koh TJ, Bryer SC, Pucci AM, Sisson TH (2005) Mice deficient in plasminogen activator inhibitor-1 have improved skeletal muscle regeneration. Am J Physiol Cell Physiol 289: C217–C223.
- 9 Krause MP, Moradi J, Nissar AA, Riddell MC, Hawke TJ (2011) Inhibition of plasminogen activator inhibitor-1 restores skeletal muscle regeneration in untreated type1 diabetic mice. Diabetes 60: 1964–1972.
- 10 Rahman FA, Krause MP (2020) PAI-1, the plasminogen system, and skeletal muscle. Int J Mol Sci 21: 7066.
- 11 Tamura Y, Kawao N, Okada K, Yano M, Okumoto K, et al. (2013) Plasminogen activator inhibitor-1 is involved in streptozotocin-induced bone loss in female mice. Diabetes 62: 3170–3179.
- 12 Mao L, Kawao N, Tamura Y, Okumoto K, Okada K, et al. (2014) Plasminogen activator inhibitor-1 is involved in impaired bone repair associated with diabetes in female mice. PLoS One 9: e92686.
- 13 Shimoide T, Kawao N, Tamura Y, Okada K, Horiuchi Y, et al. (2018) Role of macrophages and plasminogen activator inhibitor-1 in delayed bone repair in diabetic female mice. Endocrinology 159: 1875–1885.
- 14 Tamura Y, Kawao N, Yano M, Okada K, Okumoto K, et al. (2015) Role of plasminogen activator inhibitor-1 in glucocorticoid-induced diabetes and osteopenia in mice. Diabetes 64: 2194–2206.
- 15 Tamura Y, Kawao N, Shimoide T, Okada K, Matsuo O, et al. (2018) Role of plasminogen activator inhibitor-1 in glucocorticoid-induced muscle change in mice. J Bone Miner Metab 36: 148–156.
- 16 Okada K, Okamoto T, Okumoto K, Takafuji Y, Ishida M, et al. (2020) PAI-1 is involved in delayed bone repair induced by glucocorticoids in mice. Bone 134: 115310.
- 17 Iemura S, Kawao N, Okumoto K, Akagi M, Kaji H (2020) Role of irisin in androgen-deficient muscle wasting and osteopenia in mice. J Bone Miner Metab 38: 161–171.
- 18 Kawao N, Koritake A, Tatsumi K, Kaji H (2018) Roles of irisin in the linkage from muscle to bone during mechanical unloading in mice. Calcif Tissue Int 103: 24–34.
- 19 Endo T (2015) Molecular mechanisms of skeletal muscle development, regeneration, and osteogenic conversion. Bone 80: 2–13.
- 20 Schiaffino S, Reggiani C (2011) Fiber types in mammalian skeletal muscles. Phyiol Rev 91: 1447–1531.
- 21 Tamura Y, Fujito H, Kawao N, Kaji H (2017) Vitamin D deficiency aggravates diabetes-induced muscle wasting in female mice. Diabetol Int 8: 52–58.
- 22 Dadgar S, Wang Z, Johnston H, Kesari A, Nagaraju K, et al. (2014) Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J Cell Biol 207: 139–158.
- 23 Rahman FA, Angus SA, Stokes K, Karpowicz P, Krause MP (2020) Impaired ECM remodeling and macrophage activity define necrosis and regeneration following damage in aged skeletal muscle. Int J Mol Sci 21: 4575.
- 24 Nordstrom SM, Carletn SM, Carson WL, Eren M, Philips CL, et al. (2007) Transgenic over-expression of plasminogen activator inhibitor-1 results in age-dependent and gender-specific increases in bone strength and mineralization. Bone 41: 995–1004.
- 25 Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, et al. (2015) Neofunction of AVCR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci USA 112: 15438–15443.
- 26 Moritake A, Kawao N, Okada K, Tatsumi K, Ishida M, et al. (2017) Plasminogen activator inhibitor-1 deficiency enhances subchondral osteopenia after induction of osteoarthritis in mice. BMC Musculoskelet Disord 18: 392.
- 27 Takafuji Y, Tatsumi K, Ishida M, Kawao N, Okada K, et al. (2018) Plasminogen activator inhibitor-1 deficiency suppresses osteoblastic differentiation of mesenchymal stem cells in mice. J Cell Physiol 234: 9687–9697.
- 28 Barbalho SM, Prado Neto EV, De Alvares Goulart R, Bechara MD, Baisi Chagas EF, et al. (2020) Myokines: a descriptive review. J Sports Med Phys Fitness 60: 1583–1590.
- 29 Kaji H (2016) Effects of myokines on bone. Bonekey Rep 5: 826.
- 30 Zanotti S, Kalajzic I, Aguila HL, Canalis E (2014) Sex and genetic factors determine osteoblastic differentiation potential of murine bone marrow stromal cells. PLoS One 9: e86757.
- 31 Ishida M, Kawao N, Okada K, Tatsumi K, Sakai K, et al. (2018) Serpina3n, dominantly expressed in female osteoblasts, suppresses the phenotypes of differentiated osteoblasts in mice. Endocrinology 159: 3775–3790.