新薬紹介総説
新規カルシウム感知受容体作動薬ウパシカルセトナトリウム水和物(ウパシタ®静注透析用)の薬理特性と臨床試験成績
2025 年 160 巻 3 号 p. 207-219
詳細
2025 年 160 巻 3 号 p. 207-219
ウパシカルセトナトリウム水和物(ウパシカルセト)は,味覚増強の探索から派生して開発された,日本発のアミノ酸を基盤とする構造を有する新規低分子カルシウム感知受容体(CaSR)作動薬である.ウパシカルセトは,CaSRに特異的に作用し,細胞外カルシウム(Ca)存在下で活性化させることで,副甲状腺ホルモン(PTH)の分泌を抑制すると考えられる.ウパシカルセトは,非臨床試験において,二次性副甲状腺機能亢進症(SHPT)に伴う異所性石灰化,副甲状腺過形成,骨障害及び,結合部位や薬理学的特性が検討され,SHPTに伴う諸疾患の進展抑制効果,作用機序および結合様式を支持する結果が得られた.ウパシカルセトは,臨床試験において,血液透析下のSHPT患者を対象とした国内第Ⅰ/Ⅱ相試験(AJ1001試験),国内第Ⅱ相用量調整試験(AJ1002試験),国内第Ⅲ相二重盲検並行群間比較試験(AJ1004試験)及び国内第Ⅲ相長期投与試験(AJ1003試験)において有効性,安全性が確認された.ウパシカルセトは,2021年6月に「血液透析下の二次性副甲状腺機能亢進症」の効能又は効果として製造販売承認を取得し,同年8月に発売された.
Upacicalcet sodium hydrate (upacicalcet) is a novel small-molecule calcium-sensing receptor (CaSR) modulator with an amino acid structure, developed in Japan as a derivative from research into taste enhancement. Upacicalcet specifically targets CaSR and is thought to inhibit parathyroid hormone (PTH) secretion by activating the receptor in the presence of extracellular calcium (Ca). In nonclinical studies, upacicalcet was evaluated for its pharmacological properties, binding characteristics, and effects on ectopic calcification, parathyroid hyperplasia, and bone disorders associated with secondary hyperparathyroidism (SHPT). The results supported its mechanisms of action, binding mode, and efficacy in suppressing disease progression. In clinical trials, upacicalcet demonstrated efficacy and safety in patients with SHPT undergoing hemodialysis, as assessed in domestic Phase I/II trial (AJ1001 trial), Phase II trial (AJ1002 trial), Phase III placebo-controlled trial (AJ1004 trial), and Phase III long-term administration trial (AJ1003 trial). Upacicalcet was approved in June 2021 for the treatment of secondary hyperparathyroidism (SHPT) in patients undergoing hemodialysis and was launched in August of the same year.
ウパシカルセトナトリウム水和物(商品名:ウパシタ®静注透析用25,50,100,150,200,250,300 μgシリンジ,開発コード:SK-1403/AJT240,以下;ウパシカルセト)は,旧味の素製薬株式会社(現:EAファーマ株式会社,以下:味の素製薬)により見出された新規低分子化合物であり(図1),副甲状腺細胞膜上に存在するカルシウム感知受容体(calcium-sensing receptor:CaSR)に結合することにより,主として副甲状腺ホルモン(parathyroid hormone:PTH)の分泌を抑制する静注製剤のCaSR作動薬である.国内においては,2014年より株式会社三和化学研究所がウパシカルセトの開発を行い,2020年8月に製造販売承認申請し,2021年6月に「血液透析下の二次性副甲状腺機能亢進症」を効能又は効果として承認取得した.

一般名:ウパシカルセトナトリウム水和物(Upacicalcet Sodium Hydrate).化学名:Monosodium 3-({[(2S)-2-amino-2-carboxyethyl]carbamoyl}amino)-5-chloro-4-methylbenzenesulfonate hydrate.分子式:C11H13ClN3NaO6S·x H2O.分子量:373.75(脱水物として).
本稿では,新規二次性副甲状腺機能亢進症(SHPT)治療薬ウパシカルセトの薬理学的特徴および国内臨床試験成績を中心に概要を紹介する.
SHPTは,慢性腎臓病に伴う骨・ミネラル代謝異常(chronic kindey disease-mineral and bone disorder:CKD-MBD)の主な病態であり,進展したSHPTによるPTH過剰分泌は,高回転型骨病変による骨折や血管石灰化による心血管合併症を引き起こし,透析患者のQOLや生命予後を悪化させる1–3).そのため,血清PTH濃度の適切な管理は透析下におけるSHPT患者の予後改善を図る上での重要な課題のひとつに位置づけられている1).
透析下のSHPT患者におけるPTHの管理方法としてまず行うのが内科的治療であり,個々の患者の血清リン(P)及びカルシウム(Ca)の値によって活性型ビタミンD製剤(vitamin D receptor activator:VDRA)とCaSR作動薬が使い分けられている3).
CaSR作動薬登場以前は,VDRAがSHPT治療において中心的役割を担ってきたが,小腸でのCa及びPの吸収を高め,高Ca血症,高P血症の原因となることから,しばしば投与量が制限されていた.またSHPTが進行すると,副甲状腺組織が増大し,副甲状腺細胞のビタミンD受容体(vitamin D receptor:VDR)とCaSRの発現が低下し,VDRAの治療効果が得られにくくなるとの報告がある4).さらに,重症化したSHPTでは,VDRA単独での管理はしばしば困難となり,副甲状腺摘出術(parathyroidectomy)などの治療を検討することが必要となる.
CaSR作動薬は,副甲状腺細胞膜上に存在するCaSRのCa2+結合部位とは異なる場所に結合して,生理的なリガンド作用を増強する作用様式であるアロステリックモジュレーターとして作用し,Ca2+への感受性を増加させてPTH分泌・合成を抑制する.CaSR作動薬は,血清Ca及びPが低下することから,従来のVDRAでは高Ca血症,高P血症のため,治療の継続が困難であった症例でも,CaSR作動薬の併用により内科的治療を継続することができる症例が増えている5).また,CaSR作動薬によるSHPTの治療は透析患者の心血管疾患に対するシナカルセトの効果を検討した大規模臨床試験(EVOLVE試験)において,生命予後の改善や骨折リスク低減などに寄与する可能性も報告されている6,7).一方でCaSR作動薬には低Ca血症8,9)や悪心や嘔吐などの消化器症状10,11),肝機能低下患者への使用上の注意,薬物相互作用などの問題点も指摘されている12,13).
ウパシカルセト創製の起源は,新たな味覚「コク味」に関連するγ-グルタミルペプチド,特にグルタチオンの研究中に発見された化合物である.甘味,塩味,うま味等の味覚受容体は,CaSRと同じ7回膜貫通型Gタンパク質共役受容体(GPCR)のクラスCに属し,味覚を感知する受容体として機能していることが明らかとなっている14).また,グルタチオンは「コク味」を有していることが発見されており,CaSRに結合しシグナル活性を増強させることが確認されている15).このような背景のもと,味の素製薬はグルタチオンを起点に,高活性CaSR作動薬の創製研究をすすめ,ウパシカルセトが誕生した.その後,株式会社三和化学研究所が服薬負担16),上部消化管に存在するCaSRへの刺激17–20)の軽減をコンセプトにウパシカルセトの開発を行った.
CaSRは2量体を構成し,非常に長い細胞外N末端領域を持つ膜7回貫通型のGPCRであり,GiとGq/11の2種類のGタンパク質と共役している.細胞外のCa2+を感知し不活化状態のCaSRが活性化状態となるシグナル伝達としては2つある.一つは,Giタンパク質を介しアデニル酸シクラーゼを抑制し,チロシンキナーゼを活性化させることでPTH分泌を抑制する.一方,Gq/11タンパク質はプロテインキナーゼCを活性化させることで,PTH分泌を抑制し,細胞内Ca2+流入を促進する.さらにホスホリパーゼCを活性化するとイノシトール1,4,5-三リン酸(IP3)を介して小胞体内の貯蔵Ca2+を細胞質へ放出する.これらのシグナル伝達により,PTH分泌制御がなされている(図2).

CaSRは活性化状態になると,細胞内情報伝達物質IP3が放出され,その代謝物であるイノシトール-1-リン酸(IP1)が細胞内で上昇する.ヒトCaSRを発現させたHEK293T細胞に,0.0~2.0 mMのCa2+存在下でウパシカルセト又はエテルカルセチドを添加し,IP1の細胞内含有量を指標にCaSR活性作用を評価した.その結果,ウパシカルセトは,添加濃度依存的にIP1の細胞内含有量を増加させ,EC50値は26.1 nMであった.また,Ca2+が存在しない条件下ではIP1細胞内含有量をほとんど上昇させなかった.一方,エテルカルセチドは,Ca2+非存在下であってもIP1細胞内含有量を上昇させており,アゴニスト活性を示した(図3).

以上の結果から,ウパシカルセトは,Ca2+非存在下ではCaSRをほとんど活性化させることはなく,細胞外Ca2+のリガンド作用を増強させるポジティブアロステリックモジュレーター作用を有していることが確認された.
3)構造と結合部位23)ウパシカルセトの結合部位同定を進めるにあたり,構造上2つの仮説が考えられた.1つは,シナカルセト・エボカルセト同様にフェニルアラニン骨格を要することから,膜貫通領域への結合の可能性,もう一つは,ウパシカルセトがアミノ酸を起源として構造展開されたという背景を持つことから,L-トリプトファンを代表とする芳香族アミノ酸が結合する細胞外領域への結合の可能性であり,これらの仮設を検証した(図4).

ヒトCaSRを発現させたヒト胎児由来腎臓293細胞(HEK293T細胞)を用いて,放射性ラベルをしたウパシカルセトの結合がシナカルセト又はL-トリプトファンによって阻害されるかどうかを評価したところ,L-トリプトファンを添加することにより,濃度依存的にウパシカルセトとCaSRとの結合が阻害されることが明らかとなった.一方,シナカルセトはウパシカルセトとCaSRとの結合を阻害する作用は確認されなかった(図5).以上のことから,ウパシカルセトは芳香族アミノ酸同様に細胞外領域の8つのアミノ酸残基(66番目のアルギニン,70番目のトリプトファン,147番目のセリン,170番目のセリン,272番目のセリン,297番目のグルタミン,302番目のセリン,416番目のイソロイシン)と相互作用する可能性が強く示唆され,これらのアミノ酸残基との結合親和性の検証を行った.

関与するアミノ酸残基を1つずつアラニンへ置換させた変異体ヒトCaSR発現HEK293T細胞を用いて,放射性ラベルをしたウパシカルセトの結合親和性を検証した.その結果,アミノ酸ならびに芳香族アミノ酸との結合残基のアラニン変異では,野生型で見られるCaSRとウパシカルセトとの結合親和性を消失していることが認められた.以上より,上述の8つのアミノ酸残基は,変異によりウパシカルセトとの結合能が低下したことから,ウパシカルセトの結合に重要なアミノ酸残基である可能性が推察された(図6).次に,上述のウパシカルセトの結合に関与するアミノ酸残基を1つずつアラニンへ置換させた変異体ヒトCaSR発現HEK293T細胞を用いて,ウパシカルセトのCaSR結合部位と活性との関係についてIP1を指標に検討した.その結果,66番目のアルギニン,70番目のトリプトファン,147番目のセリン,170番目のセリン,297番目のグルタミン,416番目のイソロイシンをそれぞれアラニンへ置換するとCaSRのCa2+応答性が減弱した.また,アラニン置換によって減弱したCa2+応答性が,ウパシカルセトによって部分的または完全に回復することが明らかになった.

方法:ヒトCaSR発現HEK293T細胞に,11種のCaSRプラスミドを導入し100 μMウパシカルセトナトリウムを添加し,細胞内SPAビーズと結合した受容体の発光強度を測定した.*P<0.05,***P<0.001(Dunnettの多重比較),平均値 ± 標準誤差.(文献23より一部改変して引用)
ウパシカルセトと類似構造を有するL-トリプトファンは,CaSRの細胞外領域70番目のトリプトファン,145番目のトレオニン,147番目のセリン,及び170番目のセリン等の複数のアミノ酸残基と相互作用し,CaSRを不活化状態から中間活性化状態にし,さらにCa2+存在下においてCaSRを中間活化状態から活性化することが報告されている24).
ウパシカルセトも,CaSRの細胞外領域に存在するアミノ酸結合ポケットの複数の残基と相互作用することが,コンピューターを用いたin silicoドッキングシミュレーションの検討で明らかとなった.
以上のことから,ウパシカルセトはL-トリプトファン類似構造を有し,CaSRの細胞外領域70番目のトリプトファン,145番目のトレオニン,147番目のセリン,及び170番目のセリン等の複数のアミノ酸結合残基との相互作用によりCaSRを中間活性状態へシフトし,Ca2+共存化でPTH分泌抑制作用を発揮すると示唆された(図7).

透析患者のSHPTを再現すべく,腎臓全摘ラットを用いて,ウパシカルセトの有効性と安全性を既存薬と比較評価するため,血清intact PTH(iPTH)およびCa2+濃度への影響を検討した.腎臓全摘出術をラットに施し,SHPTラットを作製した.術後3日後よりウパシカルセト(0.3,3及び30 mg/kg),エテルカルセチド(0.3及び1 mg/kg)及び溶媒(0.5 w/v%生理食塩水)をそれぞれ静脈内に単回投与し,投与後24時間後と48時間後に血清intact PTH(iPTH)及び投与後24時間後に血清Ca2+濃度を測定した.病態対象群と比較してウパシカルセト投与群及びエテルカルセチド投与群の血清iPTH濃度および血清Ca2+濃度は,用量依存的に低下した(図8).

部分腎摘+溶媒,ウパシカルセト(0.3,3,30 mg/kg)又はエテルカルセチド(0.3,1 mg/kg)投与後24時間( )又は部分腎摘+溶媒,ウパシカルセト(0.3,3,30 mg/kg)又はエテルカルセチド(0.3,1 mg/kg)投与後48時間(
)又は部分腎摘+溶媒,ウパシカルセト(0.3,3,30 mg/kg)又はエテルカルセチド(0.3,1 mg/kg)投与後48時間( )のiPTHに対する作用(左図).偽手術+溶媒(
)のiPTHに対する作用(左図).偽手術+溶媒( ),部分腎摘+溶媒(
),部分腎摘+溶媒( ),ウパシカルセト(
),ウパシカルセト( )又はエテルカルセチド(
)又はエテルカルセチド( )投与後24時間のCaに対する作用(右図).平均値 ± 標準誤差(n=6).*, **, ***:P<0.05,0.01,0.001(vs. 病態対照群,Dunnettの多重比較).(文献22より一部改変して引用)
)投与後24時間のCaに対する作用(右図).平均値 ± 標準誤差(n=6).*, **, ***:P<0.05,0.01,0.001(vs. 病態対照群,Dunnettの多重比較).(文献22より一部改変して引用)
また,ウパシカルセトの薬効用量である0.3 mg/kgの100倍の用量である30 mg/kg投与であっても血清Ca2+濃度は重症低Ca血症の基準である7.5 mg/dL未満に低下することはなかった.一方,エテルカルセチドは薬効用量である0.3 mg/kgの約3.3倍の用量である1 mg/kg投与では血清Ca2+濃度は7.3 mg/dLまで減少させた.
以上の結果から,ウパシカルセトはエテルカルセチドに比べ,SHPTラットにおける血清Ca2+濃度に与える影響が少ないことが示唆された.ウパシカルセトは,SHPTラットにおいて用量依存的に血清iPTH濃度を低下させ,エテルカルセチドと同様にPTH抑制効果を示した.血清Ca2+濃度への影響において,ウパシカルセトは高用量でもCa2+濃度が生理的下限値(<7.5 mg/dL)に達しないことが確認され,エテルカルセチドに比べ低カルシウム血症のリスクが低いことが示唆された.
5)上部消化管障害に関連する評価検討(in vivo)22)以前より,CaSR作動薬は,消化管に発現するCaSRへ作用し,胃排泄能を抑制することにより,悪心や嘔吐などの消化管障害が生じることが知られている17–20).今回,ウパシカルセトが既存薬と比較して,上部消化管障害のリスクがどの程度かを評価する目的で,上部消化管障害のサロゲート指標である胃排出率を,ラットを用いてシナカルセト及びウパシカルセトで検討した.ラットにウパシカルセト(1 mg/kg及び10 mg/kg)を皮下投与,シナカルセト(30 mg/kg)若しくは溶媒(0.5%メチルセルロース溶液)を経口投与し,その投与10分後にフェノールレッド(0.05 w/v%)呈色液を投与した.フェノールレッド呈色液投与後30分が経過した段階でラットを安楽死させ,胃を取り出してフェノールレッド残存量より胃排出率を評価した.シナカルセトは,溶媒投与群に比べて胃排出率を有意に低下させた.一方,ウパシカルセトは,1 mg/kgまたは10 mg/kgを投与しても胃排出率に有意な影響は与えなかった(図9).正常ラットにおいてPTHを有意に低下させるウパシカルセトの薬効用量は0.03 mg/kgであり,その300倍以上の投与量であっても胃排出率に影響を与えなかった.

**P<0.01(vs. 偽手術+溶媒群,Student’s t-test),n=5~6,平均値 ± 標準誤差.平均値 ± 標準誤差(n=5~6).**P<0.01(vs. 正常対照群,Studentのt検定).(文献22より一部改変して引用)
以上の結果より,ウパシカルセトはシナカルセトに比べ,ラット胃排出能に影響を及ぼす可能性は少ないことが示唆された.ウパシカルセトは静脈投与されるため,消化管CaSRへの直接的な影響が少なく,胃腸障害リスクが低くなったと考えられる.
6)副甲状腺に対する作用25)アデニン誘発腎不全ラットに,ウパシカルセト(0.2及び1 mg/kg)又は溶媒(生理食塩水;正常対照群及び病態対照群)をいずれも1週間に3回,3週間反復静脈内投与した.投与開始から3週後に副甲状腺を採取し,副甲状腺重量及び副甲状腺細胞増殖数の指標であるKi-67陽性細胞数にて検討した.その結果,正常対照群と比較し,病態対照群では副甲状腺重量及び副甲状腺Ki-67陽性細胞数が有意に増加した.一方,ウパシカルセトは,いずれの投与群においても副甲状腺重量及び副甲状腺Ki-67陽性細胞数について,病態対照群との間で有意な低下が見られた(図10).

平均値 ± SE(n=11~13),***P<0.001(vs. 正常対照群,Studentのt検定),###P<0.001(vs. 病態対照群,Dunnettの多重比較).(文献25より転載)
以上の結果から,ウパシカルセトは,腎不全病態モデルで発症する副甲状腺過形成を抑制することが示された.
7)異所性石灰化に対する作用25)6)で用いたアデニン誘発腎不全ラットの胸部大動脈,心臓及び腎臓を採取し,各組織中Ca量及び石灰化病変の指標である胸部大動脈von Kossa陽性面積率にて検討した.その結果,正常対照群と比較し,病態対照群では各組織(胸部大動脈,心臓及び腎臓)中Ca量及び胸部大動脈von Kossa陽性面積率が有意に増加した.一方,ウパシカルセトは,いずれの投与群においても各組織中Ca量及び胸部大動脈von Kossa陽性面積率について,病態対照群との間で有意な低下が見られた(図11).

平均値 ± SE(n=11~14),***P<0.001(vs. 正常対照群,Studentのt検定),#, ##, ###: P<0.05,0.01,0.001(vs. 病態対照群,Dunnettの多重比較).(文献25より転載)
以上の結果から,ウパシカルセトは,腎不全病態モデルで発症する異所性石灰化の進展を抑制することが示された.
8)骨に対する作用26)アデニン誘発腎不全ラットに,ウパシカルセト(1 mg/kg)又は溶媒(生理食塩水;正常対照群及び病態対照群)をいずれも1日1回,4週間反復静脈内投与した.投与開始から4週後に右大腿骨を採取した.右大腿骨はmicro-CT法によって骨構造の指標となる皮質骨多孔率を評価した.その結果,ウパシカルセトを投与した群では,右大腿骨の皮質骨多孔率について,病態対照群との間で有意な改善が見られた(図12).

平均 ± 標準誤差(n=9~18),**P<0.01(vs. CKD-control,Dunnett’s multiple comparison test),***P<0.01(vs. CKD-control,Dunnett’s multiple comparison test).(文献26より転載)
以上の結果から,ウパシカルセトは,腎不全病態モデルで発症する骨質の悪化を改善することが明らかとなった.
維持血液透析下のSHPT患者29例に対し,ウパシカルセト(25,50,100,200,400,600及び800 μg)を単回静脈内投与したところ,血漿中において90%以上は未変化体のまま存在し,次回の透析まで血漿中ウパシカルセト濃度が維持され,投与66時間後から70時間後に血液透析を行った結果,血漿中ウパシカルセト濃度は透析直前の値より78.4~100%低下した(図13).

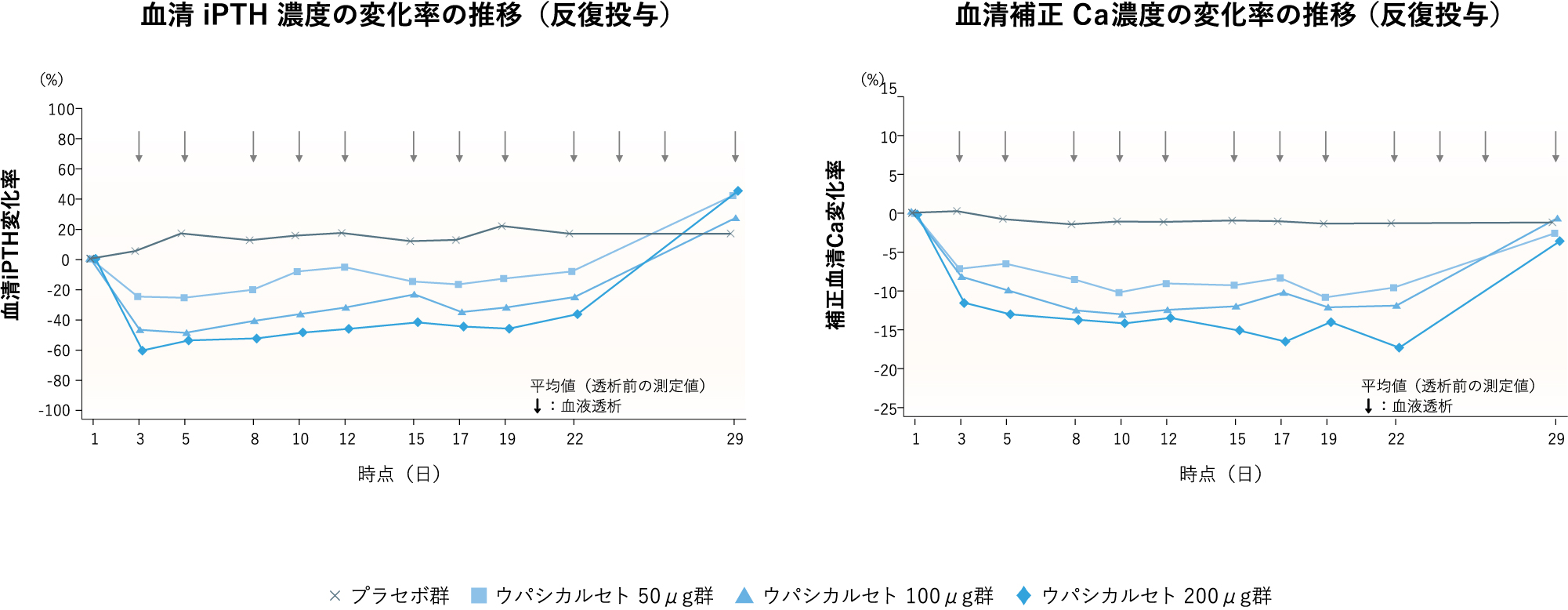
維持血液透析下のSHPT患者26例に対して,ウパシカルセト(50,100及び200 μg)を週3回,3週間,血液透析終了直前に透析回路静脈側に反復投与したところ,透析前の血漿中ウパシカルセトのトラフ濃度は上昇しないことが認められた(図14).また,ウパシカルセトの用量の増加に伴いiPTH,補正Caは低下し,最終投与後の翌週には,いずれも投与直前の値まで回復した(図15).

ヒト肝細胞を用いた検討により,ウパシカルセトが肝代謝の基質ではないことが確認された.また,主要な肝薬物代謝酵素(CYP1A2,CYP2B6,CYP2C8,CYP2C9,CYP2C19,CYP2D6,CYP2E1,CYP3A,UGT1A1,UGT2B7)に対する阻害作用や,CYP1A2,CYP2B6,およびCYP3A4の誘導作用も認められなかった.さらに,CaSR以外の60種類の受容体,イオンチャネル,トランスポーターとの結合作用を調べた結果,ウパシカルセトがこれらの標的分子に作用する可能性は認められなかった.
以上の結果から,肝機能低下による曝露量の変化や併用薬剤との薬物動態学的相互作用の懸念は低いと示唆された.
維持血液透析下のSHPT患者を対象に,ウパシカルセトの有効性及び安全性を評価した3つの国内臨床試験(用量調整試験,二重盲検並行群間比較試験,長期投与試験)の結果を紹介する(表1).
| 試験名 | 第Ⅱ相用量調整試験 (AJ1002試験)29) |
第Ⅲ相二重盲検並行群間比較試験 (AJ1004試験)30) |
第Ⅲ相長期投与試験 (AJ1003試験)31) |
|---|---|---|---|
| 対象患者 | 維持血液透析下のSHPT患者58例 | 維持血液透析下のSHPT患者153例 (ウパシカルセト群103例,プラセボ群50例) |
維持血液透析下のSHPT患者157例 |
| 試験デザイン | 多施設共同,非盲検,非対照 | 多施設共同,ランダム化,プラセボ対照,二重盲検,並行群間比較 | 多施設共同,非盲検,非対照 |
| 投与期間 | 52週間 | 24週間 | 52週間 |
| 目的 | ウパシカルセトの用量調整を行い18週間投与したときの有効性および安全性について検討した(治療第Ⅰ期). 治療第Ⅰ期を完遂した患者を対象にウパシカルセトを34週間投与したときの有効性および安全性について検討した(治療第Ⅱ期). |
ウパシカルセトの有効性について,プラセボを対照とした二重盲検並行群間比較試験にて優越性を検証し,安全性についても検討した. | ウパシカルセトを52週間投与したときの安全性及び有効性について検討した. |
| 投与例数 (完了例数) |
治療第Ⅰ期:58例(56例) 治療第Ⅱ期:56例(52例) |
ウパシカルセト群:103例(95例) プラセボ群:50例(39例) |
157例(139例) |
| 初期用量 | 治療第Ⅰ期:50 μg/回 治療第Ⅱ期:治療第Ⅰ期終了時投与量 |
血清補正Ca濃度9.0 mg/dL未満: 25 μg/回 血清補正Ca濃度9.0 mg/dL以上: 50 μg/回 |
血清補正Ca濃度9.0 mg/dL未満: 25 μg/回 血清補正Ca濃度9.0 mg/dL以上: 50 μg/回 |
| 維持用量* | 25,50,100,150,200,250, 300 μg(適宜増減) |
25,50,100,150,200,250, 300 μg(適宜増減) |
25,50,100,150,200,250, 300 μg(適宜増減) |
| 活性型ビタミンD製剤 | 治療期Ⅰ期:用法及び用量の変更又は新規投与可 禁止** 治療期Ⅱ期:用法及び用量の変更又は新規投与可 |
用法及び用量の変更又は新規投与禁止*** | 用法及び用量の変更又は新規投与可 |
| ClinicalTrial.gov number | NCT03226171 | NCT03801980 | NCT03626948 |
*iPTHが目標範囲(60~240 pg/mL)に維持されるように,投与量を調整した.増量基準は現在の用量を3週間以上維持し,血清補正Ca濃度8.4 mg/dL以上とした.減量基準は医師の判断とし,25 μg投与時の場合は休薬とした.休薬基準は補正Ca<7.5 mg/dLとし,補正Ca≧8.4 mg/dLになれば,休薬前の用量又は1段階減量した用量で再開とした.
**:症候性の低Ca血症が発現した場合は増量・新規投与,補正Ca≧11.0 mg/dLの場合は減量・休薬
***:試験薬の効果が不十分と判断された場合又は症候性の低Ca血症が発現した場合は増量・新規投与,補正Ca≧11.0 mg/dLの場合は減量・休薬
維持血液透析下のiPTH>240 pg/mLかつ補正Ca≧8.4 mg/dLのSHPT患者58例を対象に,ウパシカルセトを50 μgから投与開始し,25~300 μgの範囲で用量調整を行いながら,週3回,透析終了時の返血時に52週間投与した.その結果,主要評価項目の治療第Ⅰ期終了時である18週目のiPTHが60~240 pg/mLの範囲に達した患者の割合は57.9%,治療第Ⅱ期終了時である52週目は80.8%に達し,長期投与しても有効性が維持し,達成率の改善を継続することが確認された.
安全性において,有害事象は55例(94.8%),副作用は12例(20.7%)発現し,補正Ca減少が8例(13.8%)に認められたが,自覚症状を伴う低Ca血症はみられなかった.また,上部消化管障害として嘔吐が1例(1.7%)に認められたが,非重篤であった(表2).
| 有害事象の発現患者数 n(%) |
AJ1002試験(n=58) 52週間29) |
AJ1004試験(n=153) 24週間30) |
AJ1003試験(n=157) 52週間31) |
|
|---|---|---|---|---|
| ウパシカルセト群 (n=103) |
プラセボ群 (n=50) |
|||
| 有害事象 | 55(94.8) | 88(85.0) | 36(72.0) | 141(89.8) |
| 副作用 | 12(20.7) | 12(12) | 4(8.0) | 10(6.4) |
| 上部消化管障害 | 1(1.7) | 2(2.0) | 3(6.0) | 0(0.0) |
| 下部消化管障害 | 0(0.0) | 0(0.0) | 0(0.0) | 1(0.6) |
| Ca低下関連事象 | 8(13.8)* | 9(9.0)*** | なし | 1(0.6)*** |
| その他 | 4(6.9) | 2(2.0) | 2(4.0) | 10(6.4) |
| 重篤な有害事象 | 10(17.2) | 1(1.0)**** | 0(0.0) | 2(1.3) |
| 重篤な副作用 | 1(1.7) | 0(0.0) | 1(2.0) | 2(1.3) |
| 投与中止に至った副作用 | 1(1.7) | 0(0.0) | 2(4.0) | 2(1.3) |
| 休薬の理由となった副作用 | 7(12.1)** | 2(2.0)***** | 0(0.0) | 0(0.0) |
| 死亡に至った副作用 | 0(0.0) | 0(0.0) | 0(0.0) | 0(0.0) |
*:補正Ca減少,**:補正Ca減少(<7.5 mg/dL):7(12.1),心電図QT延長:1(1.7),***:無症候性のCa減少を補正Ca減少として集計,****:細菌性髄膜炎による死亡が1件報告されたが,試験薬との因果関係は無しと判断された,*****:補正Ca減少(<7.5 mg/dL)
本試験では,全例でウパシカルセトの初回投与量は50 μgとしたが,休薬基準である補正Ca<7.5 mg/dLとなった9例では,ベースライン時の補正Caがすべて9.0 mg/dL未満であった(表3).この結果を踏まえ,第Ⅲ相以降の試験では,ウパシカルセトの開始用量を補正Ca<9.0 mg/dLの場合には25 μg,補正Ca≧9.0 mg/dLの場合には50 μgと設定した.
| 患者 | 休薬時期 | 休薬前の ウパシカルセト 用量 |
初期値 補正Ca |
休薬判定時 補正Ca |
休薬1週間後 補正Ca |
休薬2週間後 補正Ca |
休薬3週間後 補正Ca |
休薬4週間後 補正Ca |
投与再開時 ウパシカルセト 用量 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1週目 | 50 μg | 8.9 | 7.3 | 8.4 | 8.5 | 8 | 7.7 | 休薬2週間後 (25 μg) |
| 2 | 1週目 | 50 μg | 8.8 | 7.4 | 8.5 | 9.2 | 8.5 | 8 | 休薬2週間後 (25 μg) |
| 3 | 1週目 | 50 μg | 8.9 | 7.4 | 8.4 | 8.6 | 8 | 7.8 | 休薬2週間後 (25 μg) |
| 4 | 1週目 | 50 μg | 8.7 | 7.3 | 8.5 | 8.7 | 8 | 7.7 | 休薬2週間後 (25 μg) |
| 5 | 1週目 | 50 μg | 8.4 | 7.3 | 8.7 | 8.6 | 7.8 | 7.9 | 休薬2週間後 (25 μg) |
| 6 | 1週目 | 50 μg | 8.9 | 7.4 | 8.6 | 8.7 | 7.6 | 7.7 | 休薬2週間後 (25 μg) |
| 7 | 2週目 | 50 μg | 8.4 | 7.2 | 7.9 | 8.3 | 8.2 | 投与中止 | |
| 8 | 7週目 | 25 μg | 8.7 | 7.4 | 8 | 8 | 8.8 | 9 | 休薬3週間後 (25 μg) |
| 9 | 5週目 | 50 μg | 8.4 | 7.4 | 8.1 | 8.6 | 8.1 | 8 | 休薬2週間後 (25 μg) |
| 15週目 | 50 μg | 8.4 | 7.4 | 8.5 | 7.9 | 7.9 | –b | 休薬1週間後 (25 μg) |
文献29より一部改変して引用.
維持血液透析下のiPTH>240 pg/mLかつ補正Ca≧8.4 mg/dLのSHPT患者154名を対象に,ウパシカルセトまたはプラセボを週3回,透析終了時の返血時に24週間投与した.ウパシカルセトの開始用量は補正Ca<9.0 mg/dLの場合は25 μg,補正Ca≧9.0 mg/dLの場合は50 μgより開始し,以降はiPTHが60~240 pg/mLとなるよう25~300 μgの範囲で調整した.その結果,22,23,24週時におけるiPTH平均値の管理目標値達成率は,ウパシカルセト群で67.0%,プラセボ群で8.0%であった.また,投与群間の割合の差は59.0%[95%信頼区間:47.2%,70.8%]であり,統計学的に有意であった(図16).また,ウパシカルセト群は,プラセボ群と比較して,iPTH,補正Ca及び線維芽細胞増殖因子23(FGF-23)の有意な低下が認められた.

安全性において,有害事象は,ウパシカルセト群で88例(85%)及びプラセボ群で36例(72%)に認められ(P=0.076),副作用は,ウパシカルセト群で12例(12%)及びプラセボ群で4例(8%)であり,両群間の発現率に有意な差は認められなかった(P=0.583).ウパシカルセトとの因果関係がある重篤な有害事象は認められなかった.ウパシカルセト群において,9例(9%)に低Ca血症の副作用が認められたが,すべて補正Caの低下であり,症候性の低Ca血症の発現は認められなかった.9例中2例では,補正Ca<7.5 mg/dLとなり,ウパシカルセトを休薬したが,いずれの症例も休薬1週間以内に補正Ca≧8.4 mg/dLに回復し,遷延的な補正Caの低下は認められなかった.また,上部消化管障害として,ウパシカルセト群において,悪心が1例(1.0%),食欲減退が1例(1.0%)に認められたが,いずれも非重篤であった(表2).
3)国内第Ⅲ相長期投与試験【AJ1003試験】31)維持血液透析下のiPTH>240 pg/mLかつ補正Ca≧8.4 mg/dLのSHPT患者157例を対象に,ウパシカルセトを週3回,透析終了時の返血時に52週間投与した.ウパシカルセトの開始用量は補正Ca<9.0 mg/dLの場合は25 μg,補正Ca≧9.0 mg/dLの場合は50 μgより開始し,以降はiPTHが60~240 pg/mLとなるよう25~300 μgの範囲で調整した.その結果,ウパシカルセト52週の投与により,iPTHが60~240 pg/mLの範囲に達した患者の割合は94.2%であった.iPTHの有意な低下が認められ,初期値(≦300 pg/mL,301~499 pg/mL又は≧500 pg/mL)に関わらず,iPTHの経時的な低下が観察され240 pg/mL以下に管理できた.また,補正CaおよびPの有意な低下も認められた(図17).さらには,ウパシカルセト投与により,投与前値と比較して,副甲状腺体積の有意な縮小効果が認められた.安全性においては,低Ca血症が1例(0.6%)認められたが,無症候性の補正Ca減少であり,休薬にも至らなかった.便秘が1例(0.6%)認められたが,上部消化管障害の副作用は認められなかった(表2).

ウパシカルセトは,「コク味」刺激の増強作用の探索過程からスピンアウトした,新規低分子CaSR作動薬であり,CaSRに対して複数のアミノ酸残基を介して特異的に作用する薬剤である.静注製剤とすることで,確実な投与が可能となっただけでなく,上部消化管への直接作用が軽減された.また,Ca2+非存在下ではCaSRを活性化させることはなく,細胞外Ca2+のリガンド作用を増強させるポジティブアロステリックモジュレーター作用を有しており,補正Caに応じた開始用量の選択が可能になった.さらに,CYPによる薬物相互作用リスクも軽減された.
日本において,ウパシカルセトは,「血液透析下の二次性副甲状腺機能亢進症」を効能又は効果として,2021年6月に製造販売承認され,同年8月に2剤目の静注CaSR作動薬として販売開始されている.今後,臨床現場において,既存CaSR作動薬とは異なる特性を持つウパシカルセトは,新たな治療選択肢となることが期待される.
大槻 健樹,赤利 精悟,柏木 直美,小野 嘉之(株式会社三和化学研究所).
本稿の執筆に際し,貴重なご助言を賜りました,本田 大輔,後藤 守兄,中村 敬志,保澤 寿美,米田 真司(株式会社三和化学研究所)に深く感謝申し上げます.