Original papers
Characteristics and Kinetic Properties of L-Rhamnose Isomerase from Bacillus Subtilis by Isothermal Titration Calorimetry for the Production of D-Allose
2015 Volume 21 Issue 1 Pages 13-22
Details
2015 Volume 21 Issue 1 Pages 13-22
L-Rhamnose isomerase, an aldose-ketose isomerase, is an important enzyme for rare sugar production. In this paper, L-rhamnose isomerase (L-RhI) from Bacillus subtilis 168 (B. subtilis L-RhI) was cloned and expressed in Bacillus subtilis WB600 for the production of D-allose. The obtained crude L-RhI was then purified through a HisTrap HP affinity chromatography column and an anion-exchange chromatography column. The characteristics of purified L-RhI were analyzed subsequently. The enzyme was activated in presence of Mn2+, and it had thermal advantages as its optimum temperature was 70°C, and 85% of its activity was retained after 4 hours' incubation at 60°C. In addition, isothermal titration calorimetry (ITC) was for the first time employed to determine the isomerization of rare sugars, and the kinetic data showed that B. subtilis L-RhI had relatively low Km values on the substrates of L-rhamnose, L-mannose, D-ribose and D-allose. Using D-psicose as the substrate, no by-products but only D-allose was generated catalyzed by B. subtilis L-RhI, and the yield of 37.51% was obtained when the reaction reached its equilibrium at 60°C.
Rare sugar, a class of monosaccharides, is quite minimal in nature but has unique physiological functions. It is proven to be of paramount significance not only in food industries but also in pharmaceutical and nutritional industries on account of their multipurpose applications, such as their uses as reduced calorie sweeteners, inhibitors of microbial growth, bulking agents, and memory enhancers (Bautista et al., 2000; Lawson et al., 2002; Levin et al., 1995; Livesey and Brown, 1995, 1996).
Biotransformation method is considered to have particular advantages over chemical method on many aspects, such as more facilitated processing, better activity, and higher specificity under mild conditions with a low occurrence of by-products. Izumoring strategy provided a systematic biotransformantion method of almost all rare sugars (Granström et al., 2004). In the strategy, aldehyde-ketone isomerase is the essential part of the biotransformation chain, which catalyzes the reversible reactions between aldehydes and ketones. These isomerases, however, are far from adequacy currently. Furthermore, their applications are usually limited due to strict substrate selectivity and activity.
L-Rhamnose isomerase (L-RhI, E.C. 5.3.1.14), catalyzes the reversible aldose-ketose isomerization between L-rhamnose and L-rhamnulose. As an important enzyme in Izumoring strategy, some L-RhIs can also catalyze many other reactions, and therefore has the potential for the mass production of various rare and commercially expensive sugars, such as L-talose, D-gulose, and D-allose (Bhuiyan et al., 1999; Menavuvu et al., 2006). D-Allose, a C3 epimer of D-glucose, was proven to be of potential medical and agriculture benefits, such as cancer cells-suppression (Mitani et al., 2009) and rice pathogen resistance (Kano et al., 2010). Given that the precursor D-psicose can be prepared from abundant D-fructose, D-allose production by L-RhI is considered to have application prospect in food industry (Scheme 1).

The production of D-allose using D-psicose as the substrate by P. stutzeri L-RhI
So far, L-RhI has been found to be derived from various organisms such as Aerobacter aerogenes, Escherichia coli, Lactobacillus plantarum, Mycobacterium smegmatis, and Pseudomonas stutzeri (Noltmann 1972; Badia et al., 1989; Domagk and Zech 1963; Izumori et al., 1976; Bhuiyan et al., 1999). However, only a few of them were proven to have both credible thermostability and relatively broad substrate spectrum, and D-altrose as the byproduct was inevitable in the reactions producing D-allose catalyzed by these L-RhIs. Moreover, due to the structure similarity and absence of ultraviolet absorption, rare sugars are difficult to be separated and detected using common facilities. Refractive index can only give a low sensitive detection in the micromolar range, therefore sometimes is not adequate for the kinetic study.
In the present work, L-rhamnose isomerase gene (L-RhI) from Bacillus subtilis 168 (accession number: AL009126, region: 3197933-3199207) was cloned and was then expressed in Bacillus subtilis WB600. Biochemical characteristics were investigated, and kinetic properties of the enzyme were for the first time studied by isothermal titration calorimetry. Application of B. subtilis L-RhI on the production of D-allose was also explored.
Materials L-Rhamnose, D-allose, D-psicose and other rare sugars were obtained from Kagawa University (Kagawa, Japan). Tryptone, yeast extract, agar, and the other chemicals and reagents used in this study were purchased from Sigma-Aldrich and they were of analytical grade.
Bacterial strains, plasmids, and media B. subtilis strain WB600 and plasmid pMA5 were used as the host cell and expression vector, respectively. B. subtilis cells were cultivated in Luria-Bertani (LB) medium.
Expression of L-RhI in B. subtilis The purified chromosomal DNA from B. subtilis 168 (accession number: AL009126, region: 3197933-3199207) was used as a template for PCR. To amplify the L-RhI gene, we synthesized the following primers according to the DNA sequence revealed in DNA bank: forward primer, 5′-CGCGGATCCATGACCATAAAAGCCAATTATG-3′, with an engineered BamH I site (underlined); and a universal reverse primer, 5′-ATTCGACGCGTTTAGTGGTGGTGGTGGTGGTGGACAATCGGAGAAGATG-3, with a Mls I site (underlined) and a 6×His tag at the C-terminal of the translated protein. The PCR fragment from B. subtilis 168 was digested with BamH I and Mlu I restriction enzymes, and was ligated with BamH I -Mlu I digested pMA5 vector to create pMA5-L-RhI. The recombinant plasmid was transformed into E. coli DH5α for enrichment, and was then transformed into B. subtilis strain WB600 for expression.
The recombinant cells of B. subtilis were cultivated in LB medium containing 50 µg/mL of kanamycin while being shaken at 37°C and 200 rpm for 16 h.
Purification of the recombinant L-RhI The expressed L-RhI was harvested from culture broth by centrifugation at 6,000 × g for 10 min at 4°C and then suspended in lysis buffer (20 mM Tris-HCl, 1M NaCl, and 0.5% glycerol, pH 8.0). The suspended cells were disrupted using a 2000 high-pressure homogenizer (APV, Holland) at 900 – 1,000 bars. The supernatant was obtained by centrifugation at 15,000 × g for 30 min at 4°C and filtered through a 0.45-µm filter. The crude extract was applied to a 5-mL HisTrap HP affinity chromatography column (GE Healthcare Biosciences, USA) equilibrated with binding buffer (20 mM Tris-HCl, 1M NaCl, and 0.5% glycerol, pH 8.0). The column was washed with the same buffer, and the bound protein was eluted with a linear gradient between 0 and 200 mM imidazole at a flow rate of 5 mL/min. Highly active fractions were pooled and subsequently desalted and concentrated using Amicon Ultra 15 (Millipore, USA). The resulting solution was then loaded onto a DEAE anion-exchange chromatography column (GE Healthcare Biosciences, USA) equilibrated with 30 mM HEPES buffer (pH 6.0) and eluted with a linear gradient between 0 and 500 mM NaCl at a flow rate of 1 mL/min. After desalting and concentration against 100 mM Glycine-NaOH buffer (pH 8.5), the resulting solution was used as a purified enzyme. All purification steps were carried out with an ÄKTA PurifierTM 100 system (GE Healthcare Biosciences, USA).
Effects of metal ions and temperature on L-RhI L-RhI activity was measured in a 0.5-mL reaction mixture containing 100 mM Glycine NaOH buffer (pH 8.5), 1 mM metal salts, and 50 µL of appropriately diluted enzyme. The reaction was initiated by the addition of L-rhamnose (final concentration, 1%). The mixture was then incubated for 30 min at 60°C and the reaction terminated by the addition of 50 µL trichloroacetic acid (10%). The formation of L-rhamnulose was determined spectrophotometrically following the cystein-carbazole method (Dische and Borenfreund, 1951).
To prepare the control sample without metal ions, B. subtilis L-RhI was pre-dialyzed against Na2EDTA to deionization. After dialysis against Na2EDTA, enzymes incubated with 1 mM LiCl, NaCl, KCl, CoCl2, MnCl2, NiCl2, MgCl2, BaCl2, ZnCl2, CuCl2, or FeCl3 were used to determine the effects of metal ions on L-RhI activity.
The effects of temperature on the enzymatic activity of L-RhI were determined for 30 min at various temperatures, ranging from 40 to 85°C. L-RhI was incubated at various temperatures (60, 65, 70, and 75°C) for 0, 60, 120, 180. and 240 min, and the residual activities were examined to determine the thermal stability of the enzyme.
Circular dichroism (CD) spectra Circular dichroism spectra were recorded on a Chira scan circular dichroism spectrometer (Applied Photo-physics, UK) equipped with a peltier element for temperature control. The optical path length was 1 mm. Data were recorded at 30, 35, 40, 45, 50, 55, 60, 65, 70 and 75°C. Buffer scans were recorded and subtracted for each sample spectra. The data was analyzed by GLOB3.
Equilibrium conversion analysis Isomerization of the substrate and accumulation of the products in the reaction aqueous were determined by a high-performance liquid chromatography (HPLC; Agilent, USA) using a separation column (ZORBAX Carbohydrate, Agilent) at 30°C eluted with 85% acetonitrile at a flow rate of 1.0 mL/min, and refractive index detection.
Microcalorimetry titration studies Titration calorimetry measurementswere performed with a TAM III calorimeter (TA Instruments-Waters LLC, USA). Protein solutions for isothermal titration calorimetry (ITC) were dialyzed extensively overnight against buffer (100 mM Glycine-NaOH, pH 8.5). Aldose substrate solutions of L-rhamnose, D-allose, L-mannose and D-ribose were prepared by diluting with the protein dialysate.
Heat flow (microcalories per second) was recorded as a function of time. The heat generated by dilution of the substrate was determined under the same experimental conditions except for the absence of enzyme. The method of single injection was used to determine the apparent molar enthalpy (ΔH) as follows: Subtracting the heat of dilution from the total heat evolved during the reaction and dividing by the number of substrate moles reacted in the sample cell when the reaction reached its equilibrium. The number of substrate moles reacted was calculated by the product of the number of substrate moles injected into the sample cell multiplied by the equilibrium conversion determined by HPLC. The heat involved in the assay was found by integrating the area under the heat flow curve.
There are two different ITC methods for performing an enzyme kinetic experiment: single injection and multiple injections (or continuous).
To obtain the kinetic data of D-ribose, the method of single injection was employed as follows: 27.22 µM B. subtilis L-RhI in 0.2044 mL of 100 mM Glycine buffer, pH8.5, 1 mM MnCl2 was allowed to reach thermal equilibrium at 60°C. After a 2-min wait, a single injection of 3 µL 50 g/L D-ribose was made.
To obtain the kinetic data of L-rhamnose, D-allose and L-mannose, the methods of multiple injections were implemented as follows: 0.20 µM B. subtilis L-RhI in 0.850 mL of 100 mM Glycine buffer, pH 8.5, 1 mM MnCl2 was allowed to reach thermal equilibrium at 60°C. After a 5-min wait, 12 successive injections of 3 µL 50 g/L L-rhamnose were made every 115 s; 14.27 µM B. subtilis L-RhI in 0.850 mL of 100 mM Glycine buffer, pH 8.5, 1 mM MnCl2 was allowed to reach thermal equilibrium at 60°C. After a 5-min wait, 10 successive injections of 3 µL 50 g/L D-allose were made every 180 s; 27.22 µM B. subtilis L-RhI in 0.2044 mL of 100 mM Glycine buffer, pH 8.5, 1 mM MnCl2 was allowed to reach thermal equilibrium at 60°C. After a 2-min wait, 10 successive injections of 3 µL 50 g/L L-mannose were made every 15 s.
Analyzing the kinetic ITC data The reaction rate (e.g., v0 or vmax) is typically expressed in moles of product formed per unit of time (or moles of substrate consumed per unit of time). The raw calorimetric signal is expressed as a power (e.g., µcal/sec or µJ/sec). This heat rate is simply equal to the reaction rate multiplied by the enthalpy change for the reaction. The raw calorimetric signal is thus a direct measure of the reaction rate making the calorimeter an ideal instrument for kinetic studies.
When performing a single injection ITC kinetic experiment, the substrate solution is injected into the cell containing the enzyme solution producing a heat response which eventually returns to baseline after all of the substrate has reacted. Because the reaction rate can be obtained by dividing the heat with ΔH, the heat flow can first be converted to reaction rate as a function of time, and [S] can then be obtained by substracting the area integration under the rate curve from the initial substrate concentration injected, which in the end generate a curve of reaction rate as a function of [S] to fit a Michaelis-Menton model.
The analysis methods for continuous ITC kinetic experiments are slightly different with those used for single injection kinetic experiments. As mentioned previously, if the ΔH value has been determined from a single injection experiment, v0 can be calculated using the same equation used for the single injection experiment. Each injection of the substrate generates a reaction rate and its corresponding [S], and multiple injections constitute an entire rate and [S] curve. The curve of continuous experiment is generated during the injection course, whereas that of a single injection experiment is generated during the consumption course after the injection. Therefore, the advantage of multiple experiment is that it can be used for a production-inhibited reaction since the production accumulation is rather negligible during the injection course.
Protein expression and purification B. subtilis L-RhI possesses 424 amino acids and a calculated molecular weight of 48,572 Da, which was deduced from the nucleotide sequence of the putative L-RhI gene and analyzed by the DNAMAN software.
In this study, the food-grade expression system of B. subtilis was used. The L-RhI gene was at the control of the HpaII constitutive promoter. The expressed L-RhI was in the form of a soluble enzyme. Compared to the non-expression control, extra bands of about 48 kDa were found both in the total cell lysate and in the soluble supernatant of the recombinant B. subtilis transformed with pMA5-L-RhI, as revealed by SDS-PAGE (Fig. 1(A)).

(A).SDS-PAGE analysis of L-RhI expressed in B. subtilis
M: protein marker; 1: total cell lysate obtained from a non-expression control of B. subtilis WB600 transformed with pMA5; 2: total cell lysate obtained from B. subtilis WB600 transformed with pMA5-L-RhI; 3: the soluble supernatant of B. subtilis WB600 transformed with pMA5-L-RhI; 4: the inclusion body of B. subtilis WB600 transformed with pMA5-L-RhI.
(B). Purification of the recombinant L-RhI expressed in B. subtilis.
M: protein markers; 1: the soluble supernatant of B. subtilis WB600 transformed with pMA5-L-RhI; 2: recombinant L-RhI purified through Ni column; 3: recombinant L-RhI purified through DEAE anion-exchange chromatography column.
The obtained crude protein was then purified through a HisTrap HP affinity chromatography column and a DEAE anion-exchange chromatography column. SDS-PAGE revealed a highly pure enzyme of recombinant B. subtilis L-RhI with the molecular mass of approximately 48 kDa (Fig. 1(B)), which was in agreement with the DNA sequence analysis.
Effects of metal ions and temperature of L-RhI After the treatment with EDTA, only a small amount of the enzyme activity was retained, which was then remarkably activated by 47 times in presence of 1 mM MnCl2 (Table 1). Because the enzyme activity was quite minimal in the absence of metal ions, B. subtilis L-RhI could be considered as metal ion-dependent enzyme. Co2+, Mg2+, Fe2+ could enhance the protein activity too. On the other hand, some monovalent metallic ions inhibited the enzyme, e.g. K+ and NH4+. Similar effects were also found in L-RhIs from other organisms. B. pallidus L-RhI was Mn2+ and Co2+ dependent (Poonperm et al., 2007). The crystal structure of P. stuzeri L-RhI and E. coli L-RhI revealed that they require a divalent metal ion for their activity, one or two metal ions were observed. One is “structural” (M-1) to help the substrate binding, and the other is “catalytic” (M-2) to help the hydride shift (Korndörfer et al., 2000, Leang et al., 2004a).
| Metal ions (1 mM) | Relative activity (%) |
|---|---|
| None | 100 |
| MnCl2 | 4749 |
| CoSO4 | 1043 |
| MgCl2 | 288.9 |
| FeSO4 | 113.0 |
| NiCl2 | 81.14 |
| CuSO4 | 55.41 |
| ZnCl2 | 36.45 |
| CaCl2 | 34.92 |
| (NH4)2SO4 | 32.96 |
| NaCl | 32.30 |
| BaCl2 | 32.30 |
| KCl | 31.87 |
| Na2B4O7 | 10.07 |
The temperature effect revealed that B. subtilis L-RhI worked best at 70°C(Fig. 2). The enzyme was then incubated at different temperatures for 10 hours to examine its thermal stability (Fig. 3). The activity decreased very slowly at 60°C to 85% after 4 hours, and 51.7% of the activity was still retained after 10 hours, which suggested that its half-life at 60°C was over 10 hours. When incubated at 65°C, 80% of the activity was retained after 2 hours and the half-life was about 6 hours. Although the optimal temperature was 70°C, the activity dropped with a steeper slope to a minimal level after 1 hour's incubation.

Effects of temperature on the activity of L-RhI

Thermostability of B. subtilis L-RhI
The experiments were carried out in triplicate in parallel.
Circular dichroism spectra gave another clue for the thermal stability of the enzyme. Thermal denaturation was assessed by monitoring the temperature dependence of the circular dichroism spectral signal at 200 – 280 nm. The results showed that the melting temperature (Tm) of free L-RhI was about 55.4°C in accordance with the optimal reaction temperature. Mn2+ didn't affect a lot on the thermal stability, and Tm of the Mn2+ added sample was 56.7°C(data not shown).
Substrate specificity and the production of D-allose As an aldehyde-ketone isomerase, some L-RhIs are able to react with other sugars in addition to L-rhamnose. However, the substrate specificities vary from different species. L-RhI from P. stutzeri was active with almost all the D- and L-forms of aldoses, as well as ketohexoses and ketopentoses (Leang et al., 2004a). On the other hand, E. coli L-RhI showed rather strict substrate recognition, which catalyses almost exclusively L-rhamnose and a very few of other sugars (Leang et al., 2004b).
Four aldose substrates were used to study the substrate specificity and enzyme kinetics of the recombinant B. subtilis L-RhI (Table 2). Due to the structure similarity, rare sugars are difficult to be separated. Moreover, Refractive index, as the only universal detection method, sometimes is not sensitive enough for the kinetic study. On the other hand, modern isothermal titration calorimetry make it possible to measure heat rates as small as 0.1µcal/sec, allowing for the precise determination of reaction rates in the range of 10−12 mol/sec. Values for Km and kcat, in the ranges of 10−2 – 103µM and 0.05 – 500sec−1, respectively, can be determined by this method. Therefore, isothermal titration calorimetry was, for the first time, used for the evaluation of rare sugar producing enzymes (Fig. 4).
| substrate | equilibrium conversion | ΔH (cal/mol) | Km (mM) | kcat (s−1) | kcat/Km (mM−1*s−1) |
|---|---|---|---|---|---|
| L-rhamnose | 45% | −1647.06 | 0.49 | 7.71 | 15.75 |
| L-mannose | 74% | −52.77 | 8.01 | 10.46 | 1.31 |
| D-ribose | 84% | −24.5 | 8.64 | 30.72 | 3.56 |
| D-allose | 86% | −860.83 | 5.98 | 0.74 | 0.12 |

Kinetic study of B. subtilis L-RhI by isothermal titration calorimetry
substrate: L-rhamnose; (B) substrate: D-allose; (C) substrate: L-mannose; (D) substrate: D-ribose
Because the single-injection method can only be applied in a reaction without production inhibition, multiple-injections method was used instead in reactions of which the equilibrium conversion is not higher than 80%, e.g. L-rhamnose as the substrate. The reaction of D-ribose was studied using single-injection method because its dilution heat was considerable. The results showed that the optimal substrate of B. subtilis L-RhI was L-rhamnose, as was indicated by the catalytic efficiencies (kcat/Km), followed by D-ribose, L-mannose and D-allose successively.
Since D-psicose can be obtained by D-tagatose 3-epimerase (Izumori et al., 1993) or D-psicose 3-epimerase (Mu et al., 2011) using D-fructose as the substrate, D-allose was therefore considered a prospect production by L-RhI using D-psicose as the substrate. The time course of D-allose production from D-psicose was determined at 60°C (Fig. 5). The HPLC chart showed that D-allose was the only production after 24 hours with the conversion 34.64%. The reaction reached its equilibrium after 48 hours with the conversion 37.51%, and no by-product was generated. The lack of a by-product in the reaction catalyzed by B. subtilis L-RhI granted the enzyme industrial potential for it simplifies the purification process.
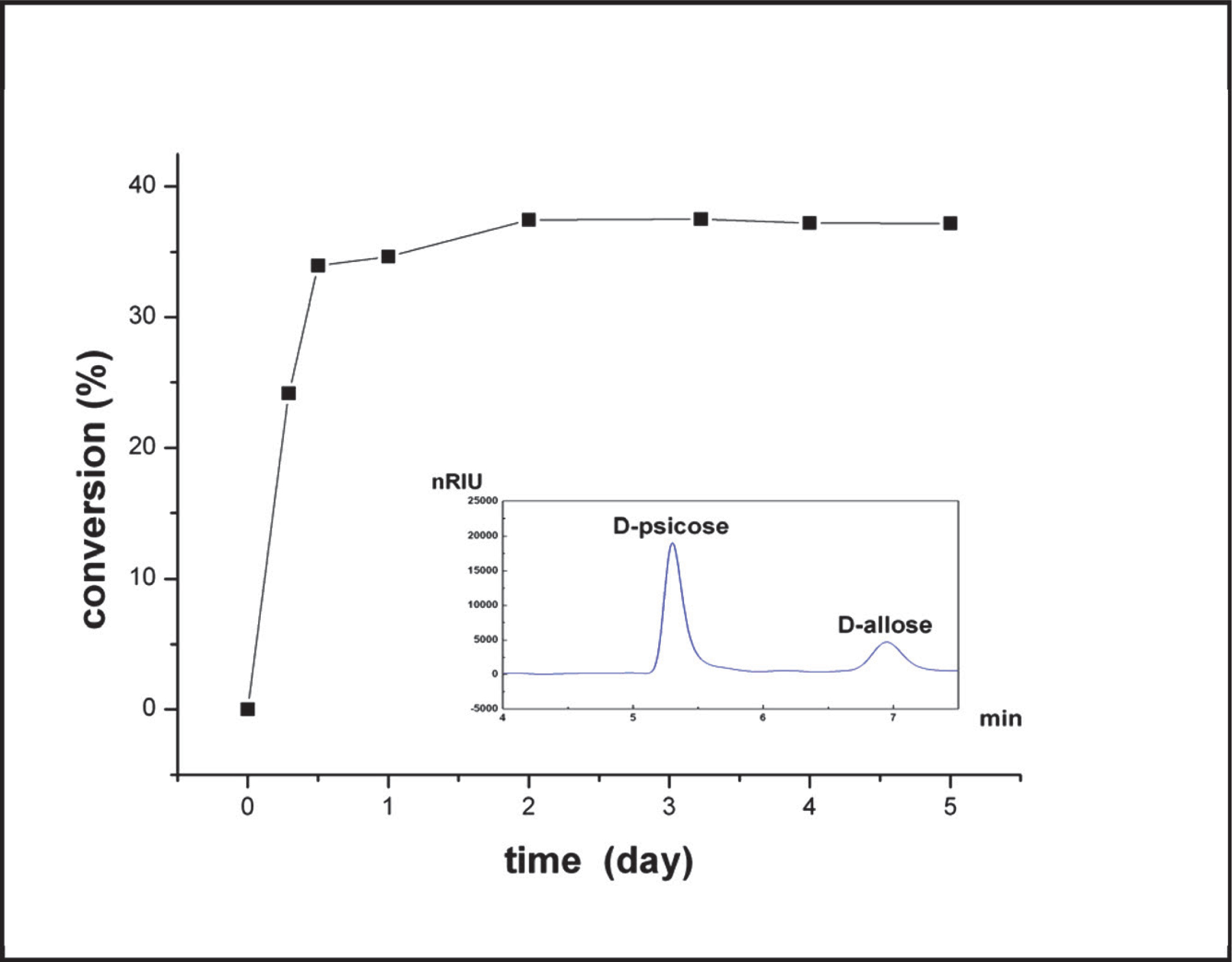
Bioconversion of D-psicose to D-allose using the purified L-RhI and the HPLC chart after 24hs' reaction
In a 0.5-mL reaction mixture containing 50 mM Glycine NaOH buffer (pH 9.0),1 mM MnCl2, and 24.5 µg of purified enzyme.The reaction was initiated by the addition of D-psicose (final concentration, 1%), and was then incubated at 60°C followed by terminationwith the addition of 50 µL trichloroacetic acid (10%).
Comparison with L-RhIs from other organisms The alignment of L-RhIs from different species revealed that B. subtilis L-RhI shares 76.42% identity with that from B. halodurans (Prabbu et al., 2011), 57.28% identity with L-RhI from E. coli K12, 56.47% identity with L-RhI from B. pallidus Y25 (Poonperm et al., 2007), 55.45% identity with L-RhI from T. saccharolyticum NTOU1 (Lin et al., 2010), but only 18.30% identity with L-RhI from P. stutzeri (Table 3). So far, crystal structures of L-RhIs from E. coli and P. stuzeris have been resolved, of which the structure of E. coli L-RhI and its relationship with xylose isomerase was the first report (Korndörfer et al., 2000). The sequence alignment of L-RhIs from E. coli and B. subtilis showed that the residues involved in the active sites of E. coli L-RhI are totally conserved (Fig. 6). In addition, five out of six putative substrate-recognizing residues of E. coli L-RhI are preserved in B. subtilis L-RhI, except for Val45. Researches revealed that Val45 together with Leu55, Ile59, Ile97, Tyr98, and Phe328 of E. coli L-RhI can form a hydrophobic area to recognize the methyl group on the C6 of L-rhamnose (Korndörfer et al., 2000). The corresponding residue in B. subtilis L-RhI is Ile43, which is also hydrophobic aliphatic amino acid and therefore would not interfere with the substrate recognition. Therefore, the alignment indicates that the substrate specificity of B. subtilis L-RhI might be similar to that of E. coli L-RhI. Further investigation revealed that the Km value of these two L-RhIs were both quite low and within a same magnitude (Table 4), which indicated that the enzymes might have a similar substrate-recognizing mechanism.
| Microorganism | Identity (%) of sequences | |||||
|---|---|---|---|---|---|---|
| B. subtilisa | B. haloduransb | E. colic | B. pallidusd | T. saccharolyticume | ||
| B. halodurans | 76.42 | |||||
| E. coli | 57.28 | 58.10 | ||||
| B. pallidus | 56.47 | 55.98 | 40.95 | |||
| T. saccharolyticum | 55.45 | 55.06 | 53.18 | 42.25 | ||
| P. stutzerif | 18.30 | 17.11 | 13.77 | 13.01 | 15.56 | |

Sequence alignment of L-RhIs from E. coli (E.coli) (accession number: WP_023281575) and B. subtilis 168 (B. subtilis) (accession number: AL009126, region: 3197933-3199207).
The alignment was performed using the DNAMAN program. Amino acid residues that are identical in all of the displayed sequences are highlighted in yellow. The residues involved in the active site and the putative substrate-recognizing residues of L-RhI from E. coli are framed in red and blue, respectively.
| aE. coli | bB. pallidus | cT. saccharolyticum | aP. stutzeri | dB. halodorans | B. subtilis | ||
|---|---|---|---|---|---|---|---|
| kcat/Km (mM−1*s−1) | L-rhamnose | ND | 13.9 | 50.8 | 14.4 | 0.28 | 15.75 |
| L-mannose | ND | 2.66 | 0.51 | 1.45 | 0.75 | 1.31 | |
| D-ribose | ND | 0.41 | 0.15 | 0.41 | ND | 3.56 | |
| D-allose | ND | 0.83 | 0.28 | 0.12 | ND | 0.12 | |
| Km(mM) | L-rhamnose | 2.0 | 4.89 | 3.53 | 11.9 | 528 | 0.49 |
| L-mannose | 5.0 | 28.9 | 58.9 | 61.7 | 119 | 8.01 | |
| D-ribose | ND | 34.9 | 148 | 38.5 | ND | 8.64 | |
| D-allose | ND | 41.8 | 121 | 42.0 | ND | 5.98 | |
| Thermal stability (°C) | 50 (10min) | 60 (1h) | 70 (2h) | 50 (10min) | 70 (25min), 60 (<15h) | 65 (6h), 60 (10h) | |
| equilibrium ratio between D-allose and D-psicose | ND | 35:65 | 29:71 | 14:75 | ND | 37.5:62.5 | |
The L-RhI from B. subtilis 168 was cloned and overexpressed in B. subtilis. The purified enzyme could be activated by Mn2+ remarkably by 47 times. The optimum temperature is 70°C, and it is stable at 60°C with its half-life over 10 hours. The conversion of D-allose by B. subtilis L-RhI was proved to be efficient at 60°C without any other by-products, and the yield of 37.51% can be obtained. The employment of ITC in this paper indicated it an efficient and prospect method for rare sugar conversions.
Acknowledgements This work was supported by National Natural Science Foundation of China (31100577), the National High Technology and Research Development Program of China (2013AA102102 and 2012AA021403) and Key Development Project of CAS (KSZD-EW-Z-019).