Original paper
Identification and Characterization of Two Novel Esterases from a Metagenomic Library
2015 Volume 21 Issue 5 Pages 649-657
Details
2015 Volume 21 Issue 5 Pages 649-657
Esterases are biocatalysts in food industry aimed for nutrition improvements, formation of flavor, and food fermentation. Two esterases EstGX1 and EstGX2 were identified based on function-based screening of a soil metagenomic cosmid library. Enzyme properties including optimum pH, optimal temperature, tolerance to organic solvents and metal ions were measured, respectively. The activity of EstGX2 could maintain about 40% after incubated at 99°C for 55 min, and could be increased in presence of 15% ethanol. The unique properties of EstGX2, high thermostability and stability in the presence of several organic solvents, may make it a promising enzyme candidate in food industry.
Esterases (EC 3.1.1.1) and lipases (EC 3.1.1.3) are the two major families of lipolytic enzymes, principally catalyze the hydrolysis and synthesis of ester compounds (Park et al., 2007), but typically differ in their substrate preference. It is widely recognized that, typically, lipases act on water-insoluble triglycerides (chain length of > 10 carbon atoms), while esterases hydrolyze short-chain (chain length of < 10 carbon atoms) esters (Rhee et al., 2004). Esterases are wild useful biocatalysts in food industry including nutrition improvements (Yang et al., 2013), formation of flavor (Wolf et al., 2009), and food fermentation (Esteban-Torres et al., 2014). Esterases can be found in plants, animals and microorganisms, and most industrial esterases are of microbial origin. Microorganisms have been a major source of novel enzymes (Torsvik et al., 2002; Strohl, 2000); However, most of the microorganism derived enzymes could not been studied from their natural hosts, because most microorganisms (> 99%) are not readily cultured in the lab, and these vast major uncultured microorganisms represent a large gene pool for biomolecules exploitation (Ferrer et al., 2005; Amann et al., 1995). Fortunately, it is possible to extract microbial DNA directly from an environmental sample and clone these DNA into easily cultured bacteria, which been termed ‘metagenomics’ (Handelsman, 2004; Daniel, 2005). A variety of enzymes have been identified using metagenomic technology, including xylanases (Qian et al., 2015), glycosyl hydrolases (Sathya et al., 2014), and amylases (Vester et al., 2015).
In this study, we constructed a metagenomic cosmid library from a chestnut grove soil sample in Yunnan, China. Two novel esterases were identified by a function-based screening assay from the library, followed by the biochemical characterization of the purified enzymes. EstGX2 with high thermostability and stability in the presence of several organic solvents might be a promising enzyme candidate in food industry.
DNA extraction and construction of metagenomic library Soil samples were collected from 20 cm below the surface of a chestnut grove in March 2013 in Yunnan (N25°20′, E102°30′). The metagenomic DNA was isolated using published protocol (Brady, 2007). Briefly, after sifting to remove large particulates, soil was heated (70°C) in lysis buffer (100 mM Tris-HCl, 100 mM EDTA, 1.5 M NaCl, 1% [w/v] cetyl trimethyl ammonium bromide, 2% [w/v] SDS, pH 8.0) for 2 h. Soil particulates were removed from the crude lysate by centrifugation, and environmental DNA (eDNA) was precipitated from the resulting supernatant with the addition of 0.7 vol isopropanol. Crude eDNA was collected by centrifugation, washed with 70% ethanol, and resuspended in TE buffer (10 mM Tris and 1 mM EDTA, pH 8.0). High molecular weight DNA was purified from the crude extract by gel electrophoresis (1% agarose, 16 h, 20 V), blunt-ended, ligated into precut pWEB-TNC (Epicentre, Charlotte, USA), packaged into lambda phage (Epicentre, Charlotte, USA) and transfected into Escherichia coli EPI100-T1R (Epicentre, Charlotte, USA).
Functional-based screening for lipolytic activity Approximately 400 – 500 library colonies were plated onto LB screening agar plates (12.5 µg/mL chloramphenicol, Generay Biotechnology, Shanghai, China; 100 µg/mL ampicillin, Generay Biotechnology, Shanghai, China; 1% [w/v] tributyrin, Acros Organics USA, Belgium, USA; and 1% [w/v] gum arabic (Liu et al., 2009), Sinopharm Chemical Reagent Co., Ltd, Shanghai, China) to find lipolytic active transformants. The plates were incubated at 37°C for 3 – 5 days, and the positive clone with a clear halo around it was singled out for latter part of subcloning experiments. The cosmid DNA were isolated from the positive lipolytic clones and transferred into E. coli DH5α for retesting the activity to avoid false positive clones.
Subcloning and DNA sequence analysis DNA of positive clones were purified and partially digested with Sau3AI (1:1000) (TaKaRa, Dalian, China), the digested DNA of 1 – 4 kb was then ligated with the pWEB-TNC vector, transformed into E. coli EPI100-T1R, and plated onto LB screening agar plates for positive subclones. The resulting positive recombinant clones were then sequenced. Sequence analysis was carried out with the BLASTx program (http://blast.ncbi.nlm.nih.gov). Furthermore, multiple sequence alignment was performed with the ClustalW2.0 program and phylogenetic analysis (Neighbor-joining tree) was done with MEGA6.0. The sequences reported in this paper have been deposited with GenBank database under accession numbers KP771883 (estGX1) and KP771884 (estGX2).
Construction of expression recombinant plasmids The putative esterase genes estGX1 and estGX2 were amplified by PCR with primer pairs of estGX1-F (5′-CATGCCATGGCGAGTCCGCAACTACAA AC-3′)/ estGX1-R (5′-CCGCTCGAGGGAAGTATGCTCGCGGA TGA-3′) and estGX2-F (5′-CATGCCATGGGCATGCCCCTGCATCCCCAG)/ estGX2-R (5′-CCGCTCGAGCTTGCGCAGCTGTTCCTT-3′), respectively. Underlining is NcoI or XhoI restriction site and the start codons are written in bold. The PCR fragments were digested with NcoI (TaKaRa, Dalian, China) and XhoI (TaKaRa, Dalian, China), and then ligated into pET-28a (+) vector that had been digested with the same restriction enzymes. The recombinant DNA was transformed into E. coli BL21 (DE3) for overexpression.
Expression and purification of EstGX1 and EstGX2 Transformants were cultured in LB broth containing 50 µg/mL kanamycin (Melonepharma, Dalian, China) at 37°C. Overnight cultures were used to inoculate 400 mL cultures of LB (1:1000 dilution), which were grown at 37°C until the OD600 reached 0.6. The temperature was then reduced to 18°C and protein expression was induced with IPTG (0.4 mM) for an additional 18 h. The cells pellet were harvested by centrifugation, resuspended in lysis buffer (50 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1% Triton X-100), and disrupted by sonication, then the crude cell lysate was withdrawn after centrifugation at 15,000 rpm for 15 min and applied to a nickel column containing His-Binding-Resin (Bio-Rad, Hercules, USA). After washing with 10 column volumes of the washing buffer (50 mM Tris-HCl [pH 8.0], 100 mM NaCl), the bound proteins were eluted with elution buffer (50 mM Tris-HCl [pH 8.0], 100 mM NaCl, 500 mM imidazole). Protein concentrations were determined with the Bradford assay using bovine serum albumin (Generay Biotechnology, Shanghai, China) as standard. Aliquots were then flash frozen and stored at −80°C. Purified protein was electrophoresed on a 12% (w/v) SDS-PAGE gel.
Biochemical characterization of EstGX1 and EstGX2 Unless otherwise described, activities of EstGX1 and EstGX2 were measured at 46.4°C and 50°C, respectively, with 0.5 mM p-nitrophenol esters as the standard substrate (Schmidt-Dannert et al., 1994) in 50 mM Tris-HCl (pH 9.0). After preincubation of the reaction mix (95 µL) for 1 min, the reaction was initiated by adding 5 µL esterase, and incubated for 5 min. The reaction was then stopped by adding 20 µL of 10% SDS. The amount of p-nitrophenol released by enzymatic hydrolysis was measured at 405 nm by Spectrophotometry (Hu et al., 2012). Blank reaction was needed in every measurement for subtracting the value of non-enzymatic hydrolysis of substrate. One unit (U) of enzyme activity was defined as the amount of enzyme required for the release of 1 µmol of p-nitrophenol per min.
To determine the substrate specificity, p-nitrophenol butyrate (C4) (Sigma, San Francisco, USA) and p-nitrophenol myristate (C14) (Sigma, San Francisco, USA) were used as substrates in the assays. A final concentration of 0.038 mM Triton X-100 was added to the reaction system to promote dissolution of p-nitrophenol myristate (Choo et al., 1998). The optimum temperature of the enzyme reaction was determined by measuring the activity at temperatures between 4°C and 60°C in Tris-HCl buffer (pH 9.0) using p-nitrophenyl butyrate as substrate. The optimum pH was determined at the optimum temperature in different buffers: 50 mM sodium citrate buffer (pH 3.0 – 6.0), 50 mM phosphate buffer (pH 6.0 – 8.0), 50 mM Tris-HCl buffer solution (pH 8.0 – 10.0). Besides, the effects on esterase activity at a final concentration of 5 mM metal ions (MnCl2, MgCl2, CaCl2, CuCl2, ZnCl2, CoCl2, and NiCl2) and metal ion chelator EDTA were detected. The detergent effects were detected for 1% (v/v) of SDS, Triton X-100, Tween-20, and Tween-80. The effects of organic solvents, including methanol, ethanol, isopropanol, acetonitrile and DMSO, were also detected at a final concentration of 15% (v/v) and 30% (v/v). All experiments were carried out in triplicate under standard experimental conditions contract to blank action. Esterase activity measured under the same conditions without additives was defined as 100%.
Construction and screening of the metagenomic library A metagenomic library was constructed with the environmental DNA isolated from a soil sample derived from a chestnut grove in Yunnan. About 1 × 105 individual cosmid clones were obtained, with a range of insert size of 33 – 45 kb. Library clones were spreaded on the lipolytic-screening LB agar plates containing tributyrin, and two cosmid clones were obtained on the basis of the formation of hydrolysis zone. Then, these cosmids were re-transformed into E. coli DH5α, using the screening plates to identified and verified as positive clones. Finally, two genes encoding putative lipolytic enzymes were identified by subcloning and subsequent sequencing.
As sequence information is not required before screening, function-driven approach is a powerful strategy that bears the potential to discover entirely novel full-length lipolytic genes and functional gene products (Daniel, 2005; Simon et al., 2009). The screening for genes exhibiting lipolytic activity was based on the ability of library clones to hydrolyze tributyrin and form hydrolysis halos when grown on agar medium containing tributyrin. This function-based screen has been used to identify lipolytic enzymes from diverse environments such as soils (Lammle et al., 2007; Elend et al., 2006; Heath et al., 2009), leachates (Rashamuse et al., 2009), sediments (Lee et al., 2006; Jeon et al., 2009), activated sludge (Roh et al., 2008), surface seawater (Chu et al., 2008) and pond water (Ranjan et al., 2005). Here, we used a function-driven approach to isolate genes encoding lipolytic enzymes, implying that a simple activity-based screening system is a useful method for the isolation of diverse genes conferring the targeted reaction in soil-based metagenomic libraries.
Sequence analysis Two putative lipolytic gene, named estGX1 and estGX2, were identified. Sequence analysis showed that estGX1 and estGX2 genes encoded putative proteins composed of 296 and 312 amino acids, respectively. BLASTx analysis showed that the deduced proteins displayed a certain degree of similarity with the known lipases/ esterases. EstGX1 was found having the highest identity of 67% with the esterase (ACL67845.1) recovered from a marine sediment-derived metagenomic library (Hu et al., 2010), while EstGX2 was observed to hold the highest identity of 57% with an esterase from Nevskia ramosa (WP_022977707.1). To a large extent, EstGX1 and EstGX2 were deduced to share the typical α/β hydrolase fold manner of many hydrolase enzymes with most lipases/ esterases.
Microbial lipolytic enzymes were divided into eight different families (I–VIII) according to the homology of amino acid sequences and properties of physiology (Arpigny et al., 1999). Phylogenetic placement of the predicted proteins EstGX1 and EstGX2 were grouped into family IV bacterial lipolytic enzymes (Fig. 1), a family displaying a striking amino acid sequence similarity to the mammalian hormone-sensitive lipase (HSL). Lipolytic enzymes of family IV were also predominantly recovered in similar function-based screens of other metagenomic libraries derived from a variety of environments such as forest soil (Lee et al., 2004; Hong et al., 2007), deep sea sediment (Hu et al., 2010).
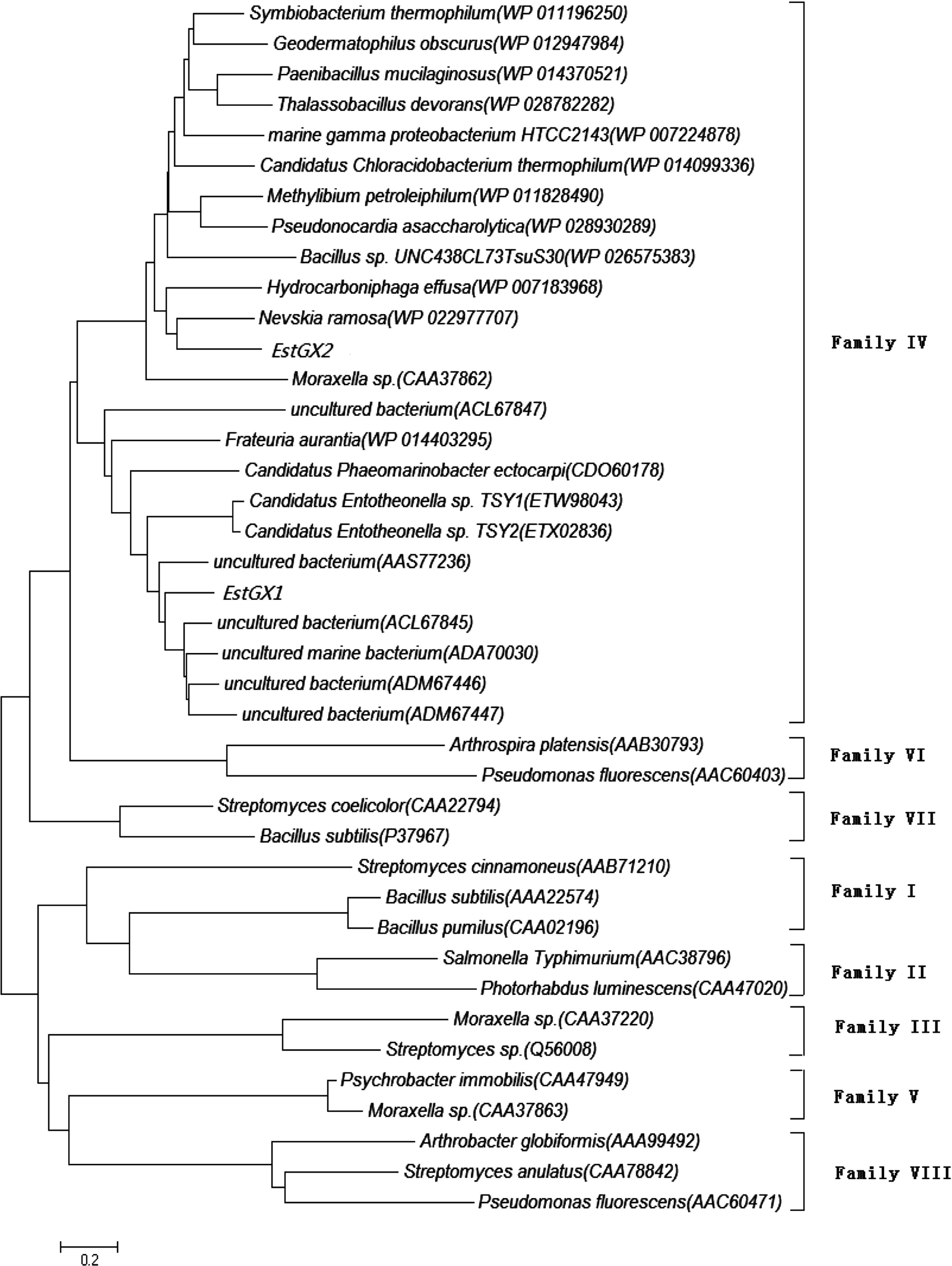
Neighbour-joining analysis of EstGX1, EstGX2 and lipolytic enzymes belonging to different families. The Poisson correction method was used to calculate evolutionary distances. Database accession numbers of other closely related lipolytic enzymes are shown in brackets after each enzyme.
Multiple-sequence alignments with representative members of family IV exhibited that EstGX2 harbored the conserved catalytic triad residues Asp253, His283 and the catalytic nucleophile Ser57 in the highly consensus pentapeptide G-X-S-X-G (Fig. 2B), which was appearing to be one of the vital active sites (Kim et al., 2004). As for EstGX1, there were the classical residues Ser144 and His268 as well, but the Asp in motif DPLR was replaced by Glu238 (Fig. 2A). The highly conserved motif HGGG, which is involved in hydrogen bonding interactions and playing a catalytic role (Fischer et al., 2003; Wei et al., 1999), is present in both EstGX1 and EstGX2 (Fig. 2A–B).

Multiple-sequence alignments of EstGX1 (A) and EstGX2 (B) with other microbial lipases/ esterases. Regions of high identity or similarity among sequences are shown as asterisks.
Over-expression and biochemical analysis of recombinant enzymes EstGX1 and EstGX2 were expressed using pET-28a (+) vector in E. coli BL21 (DE3). Both recombinant proteins were purified from the culture supernatants by nickel column chromatography. The SDS-PAGE analysis illustrated that the purified proteins showing almost a single belt possessed corresponding estimated molecular weight of 31 kDa and 33 kDa (Fig. 3), which corrected well with the predicted His-tag fusing sizes of EstGX1 and EstGX2, respectively. As detected by the Bradford method, protein content of EstGX1 was 60.66 mg/mL and 67.73 mg/mL for EstGX2.

SDS-PAGE of overexpressed recombinant EstGX1 (A) and EstGX2 (B). Lane M: protein molecular weight marker; lane 1: extract of induced E. coli BL21 (DE3) cells transformed with pET-28a (+); lane 2: total cell extract of un-induced bacteria overexpressing esterase; lane 3: total cell extract of induced bacteria overexpressing esterase; lane 4 and 5: insoluble and soluble fractions of the induced cell extract, respectively; lane 6: purified protein.
In the determination assay of suitable substrate, compared to the significant hydrolytic activity towards p-nitrophenyl butyrate (C4), EstGX1 and EstGX2 almost had no activity on p-nitrophenyl myristate (C14) (Fig. 4), indicating that EstGX1 and EstGX2 were esterases instead of lipases. The specific activity of EstGX1 and EstGX2 towards C4 were 61.56 ± 0.01 U/mg and 0.68 ± 0.01 U/mg, respectively. The specific activity of EstGX1 and EstGX2 towards C14 were 0.27 ± 0.01 U/mg and 0.08 ± 0.01 U/mg, respectively. EstGX1 and EstGX2 were all active at pH 7.0 to 10.0, while their optimum pH were both alkaline pH 9.0. However, in extremely acidic or alkaline (> pH 10.0) condition, the two enzymes would lose the ability to hydrolyze p-nitrophenol butyrate (Fig. 5A–B). Therefore, we chose p-nitrophenol butyrate as the standard substrate and pH 9.0 for subsequent assay of lipolytic activity.
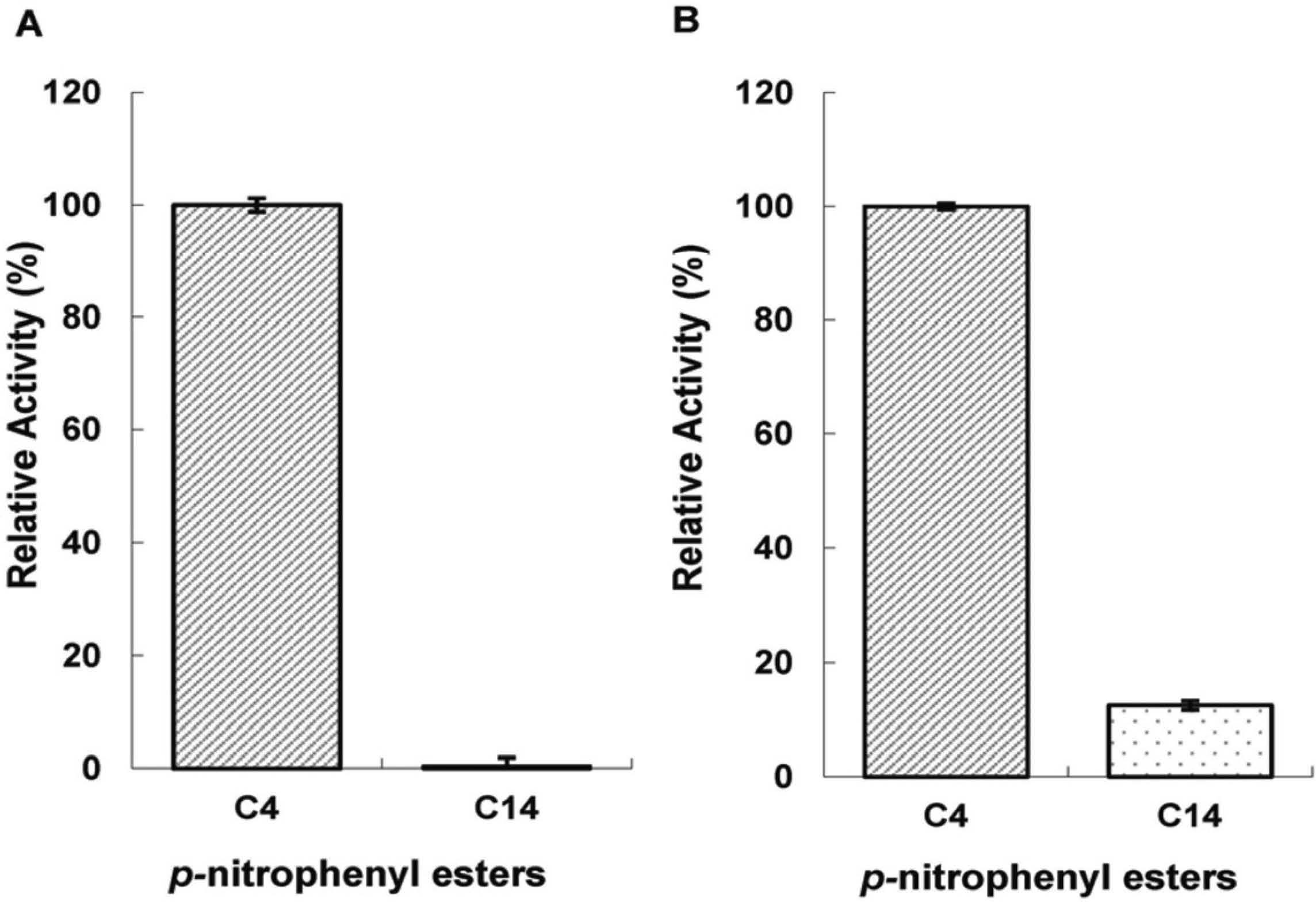
Substrate specificity of EstGX1 (A) and EstGX2 (B) toward different p-nitrophenyl esters. The relative activity of the enzyme toward p-nitrophenyl butyrate was defined as 100%.
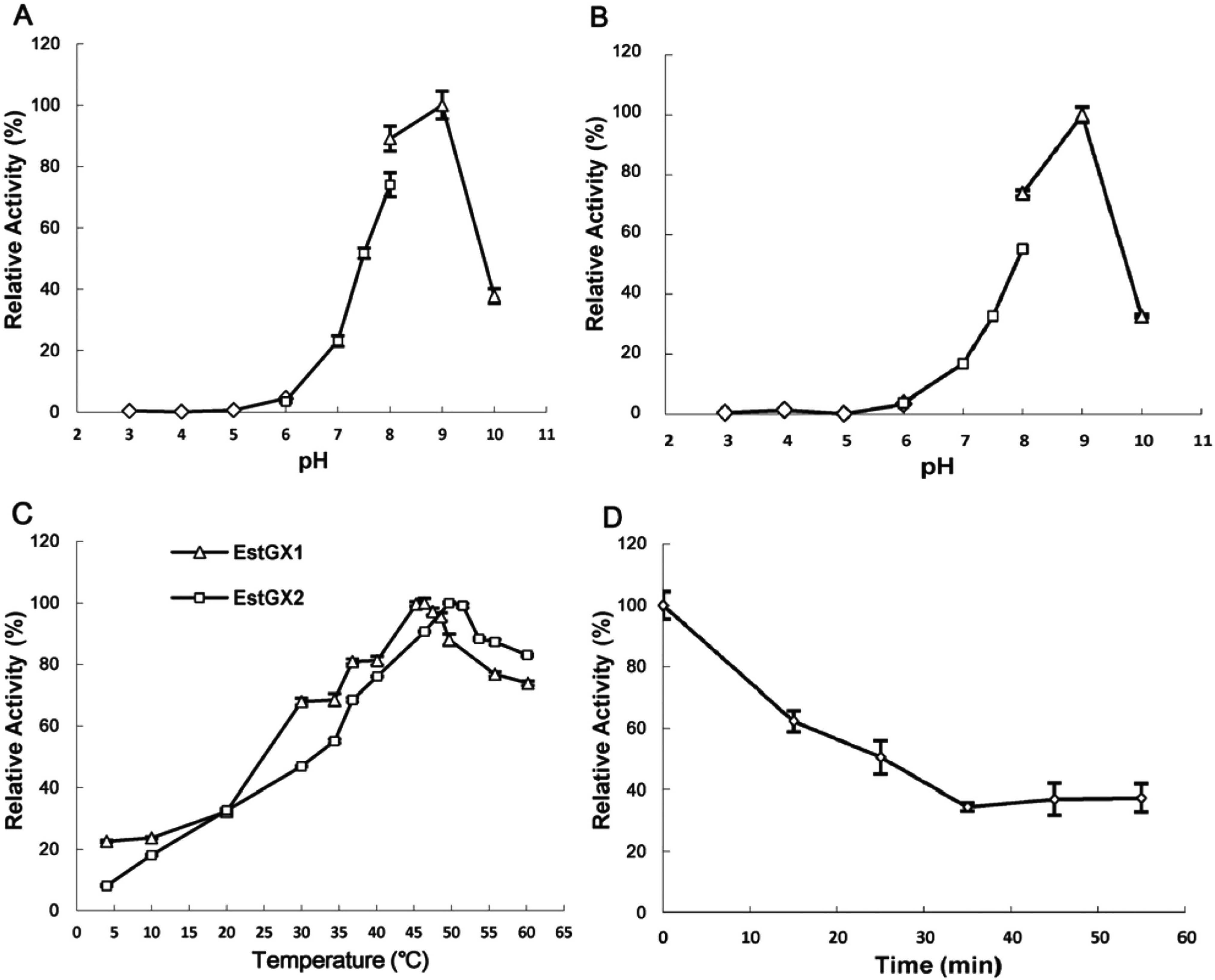
Effect of temperature and pH on the activity of EstGX1 and EstGX2. All the assays were performed for 5 min with 0.5 mM p-nitrophenyl butyrate as the substrate. Effect of pH on the activity of EstGX1 (A) and EstGX2 (B). (A–B) The assay was performed in different buffers: 50 mM sodium citrate buffer (pH 3.0 – 6.0, hollow diamonds), 50 mM sodium phosphate buffer (pH 6.0 – 8.0 hollow squares), and 50 mM Tris-HCl buffer (pH 8.0 – 10.0, hollow triangles). (C) Enzyme activities were measured over various temperatures (4 – 60°C) in 50 mM Tris-HCl buffer (pH 9.0). (D) The thermostability of EstGX2 was measured after incubating the enzyme for 15, 25, 35, 45, and 55 min in individual tubes at 99°C. The initial EstGX2 hydrolysis activity was shown as 100%.
The optimal temperature of EstGX1 and EstGX2 were 46.4°C and 50°C (Fig. 5C), respectively. It is noteworthy that the two enzymes would still maintain higher than 60% and 80% relative activity at 60°C. More interestingly, EstGX2 could retain about 40% of activity after incubation at 99°C for 55 min (Fig. 5D), while EstGX1 lost activity after incubation at 99°C for 10 min. On the other hand, EstGX1 could hold above 20% activity at 4°C, while EstGX2 showed only 8% relative activity at 4°C. Both cold-active and hot-active esterases are important for industrial applications, suggesting the two esterases here have potential for many biotechnological usage.
Furthermore, we examined the effects of different metal ions on the esterase activity at optimal temperature and pH (Fig. 6). We found that the lipolytic activity of EstGX1 was inhibited by 5 mM Mn2+, Ni2+, Cu2+, and especially Zn2+ and Co2+; whereas, the activity of EstGX2 was decreased by Cu2+, Co2+, Ni2+, Mn2+ to some extent, and almost inhibited by Zn2+ and Ca2+. Besides, metal ion chelator EDTA just had a slight effect on the esterase activity of EstGX1 and EstGX2, suggesting EstGX1 and EstGX2 may not be metalloproteins.
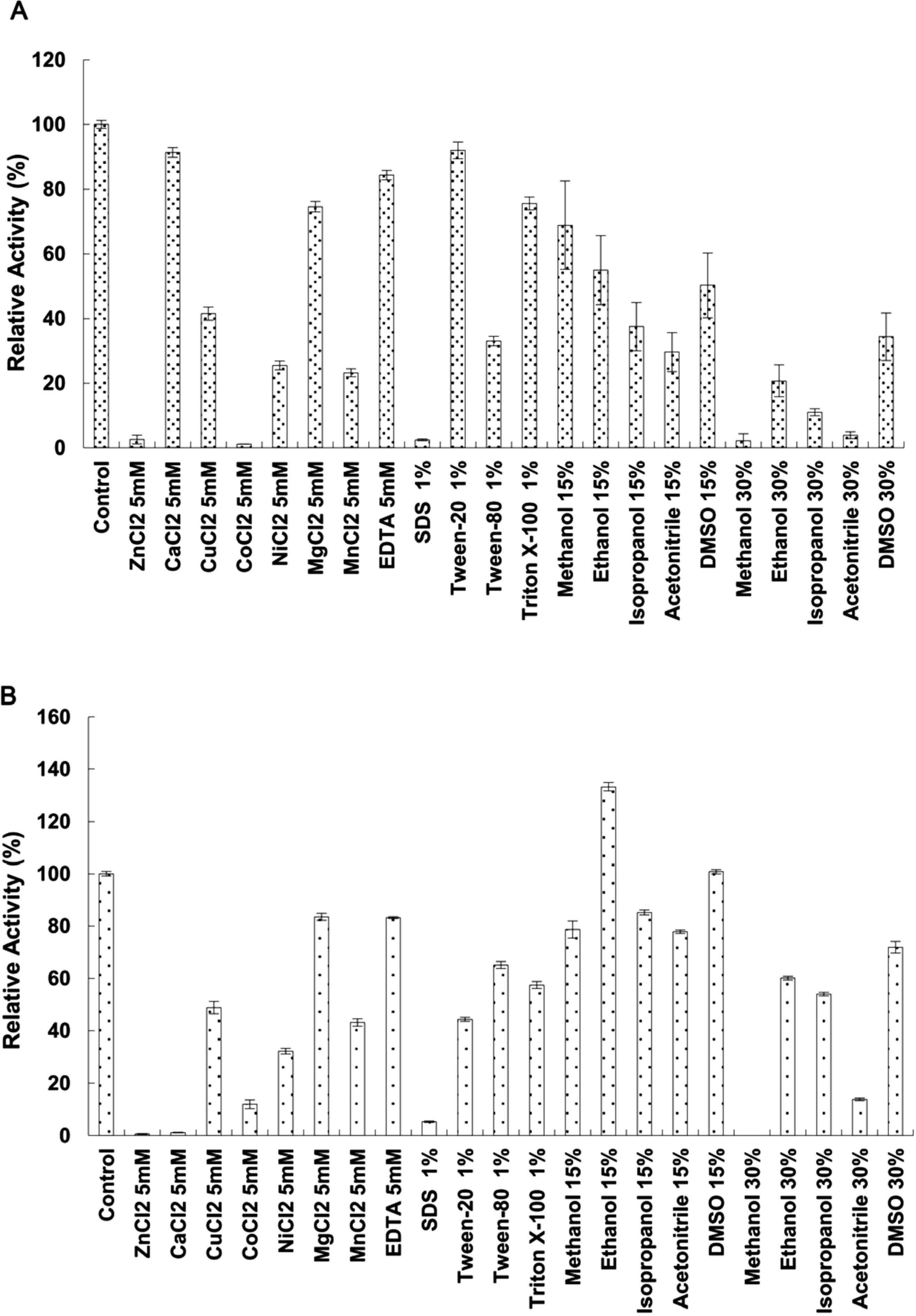
Effects of various additives on the lipolytic activity of EstGX1 (A) and EstGX2 (B). The hydrolysis rate of p-nitrophenyl butyrate was measured at the optimum temperature in 50 mM Tris-HCl buffer (pH 9.0) with 0.5 mM p-nitrophenyl butyrate as the substrate. Enzyme activity in the absence of additives was shown as 100% (Control).
Esterase tolerance towards detergent as well as organic solvent was determined at the optimum temperature in 50 mM Tris-HCl buffer (pH 9.0). The activities of two putative esterases were inhibited in the presence of 1% Triton X-100, 1% Tween-20, and especially in the presence of 1% SDS (Fig. 6). In terms of organic solvent tolerance, EstGX2 showed more tolerance than EstGX1 against most organic solvents. The enzyme activity of EstGX2 was stable in presence of 15% organic solvents, including acetonitrile, isopropanol, methanol, and even had an increase in presence of ethanol, whereas the enzyme active of EstGX1 was inhibited in presence of 15% organic solvents, including acetonitrile, isopropanol. The activities of both EstGX1 and EstGX2 were inhibited in presence of 30% organic solvent, especially in presence of methanol, isopropanol, acetonitrile for EstGX1 and in presence of methanol, acetonitrile for EstGX2.
In conclusion, two novel esterase genes, named estGX1 and estGX2, were isolated from a soil metagenomic cosmid library derived from a chestnut grove sample in Yunnan. Multiple sequence alignments and phylogenetic tree analysis revealed that both predicted enzymes were new members of bacterial lipolytic family IV. Characterization of the enzymes showed EstGX2 to be tolerant to high temperature and several organic solvents. These features of EstGX2 indicated it might be a promising enzyme candidate in food industry.
Acknowledgements This work was financially supported in part by National Natural Science Foundation of China (31370088), State key lab of Microbial Resources, Institute of Microbiology, CAS (SKLMR-20130602), and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.