Original papers
Yeast diversity investigation of ‘Beihong’ (V. vinifera × V. amurensis) during spontaneous fermentation from Guiyang region, Guizhou, China
2021 Volume 27 Issue 6 Pages 887-896
Details
2021 Volume 27 Issue 6 Pages 887-896
‘Beihong’ is a new eurasian vinifera hybrid cultivar and widely planted in many wine regions and selected for wine making in China. However, studies on the fungal diversity of this cultivar was scarce. In this study, the indigenous yeast diversity and theirs dynamic changes during the spontaneous fermentation of ‘Beihong’ from Guiyang region, Guizhou, China, was analyzed and monitored using high-throughput sequencing and culture-dependent approaches. The result demonstrated that the composition of yeast species revealed by the two methods was quite different. In addition, non-Saccharomyces yeasts were predominant in the initial stages of fermentation and then rapidly decreased, while Saccharomyces yeast sharply increased during the process. Hanseniaspora sp. and Saccharomyces cerevisiae were the main yeast isolates at the third day of fermentation. Taken together, the present study was a first step in exploring untapped yeast resources of ‘Beihong’ from Guiyang region, showing the basic information on indigenous yeast diversity, which was helpful to screen these native yeasts for producing wines with regional characteristics.
The spontaneous fermentation of wine is a complex microbial interaction process, and bacteria, yeasts and other fungi involved in this process (Li et al., 2018). Yeasts play a fundamental role, and can divide into Saccharomyces yeasts and non-Saccharomyces yeasts according to their fermentation characteristics (Jolly et al., 2014). The main role of Saccharomyces yeasts was converting sugar into ethanol, making the wine with a certain alcohol degree, and they dominated in the middle and late stages of spontaneous fermentation, while non-Saccharomyces yeasts were found in the early stages, and produced volatile and non-volatile constituents, which enhanced the flavor and sensory characteristics of wine (Morata et al., 2014; Bellut and Arendt, 2019). In brief, the sensory qualities of the wine depend on Saccharomyces' and non-Saccharomyces yeasts' interactions. Therefore, clarification the yeast population diversity and theirs' dynamics during spontaneous fermentation were helpful for selecting potential indigenous yeast strains to manufacture territorial unique wines (Wei et al., 2019). However, the microbiota remains unclear for some minority grape varieties.
China is rich in native Vitis resources including 40 species, 1 subspecies and 13 varieties, such as Vitis Amurensis, Vitis Davidii and Vitis Heyneana (Wang et al., 2019). ‘Beihong’ is a vinifera hybrid cultivar with high resistance to cold and fungus diseases using ‘Muscat Hamburg’ (Vitis vinifera L.) as the female parent and wild Vitis amurensis as the male parent by Institute of Botany (Chinese Academy of Sciences), and was approved as a new grape cultivar in China in 2008 (Wang et al., 2016; Wang et al., 2015). At present, ‘Beihong’ is planted in most vineyard regions of China. The wine produced with ‘Beihong’ is dark ruby red in color, and the wine body is clear, transparent, full-bodied and balanced (Fan et al., 2009). However, researches on ‘Beihong’ mainly focused on theirs cultivation and agronomic traits. It is well known that grape harbors various indigenous yeasts which are associated with wine quality and flavor (Wang and Liu, 2013). Therefore, it is very necessary to elucidate the diversity of indigenous yeast for wine grape varieties.
Development of molecular biotechnology provided effective tools for analyzing yeast diversity during spontaneous fermentations (Zhang et al., 2019; Ding et al., 2019). Nowadays, high-throughput sequencing technology has been widely used to analyze the diversity of grape microbial resources and the dynamic changes therein. Yeast communities collected during the spontaneous fermentations of two grape varieties of Vitis davidii Föex were examined using culture-dependent and high-throughput sequencing approaches in Wang's study, with Hanseniaspora uvarum, Pichia terricola, Saccharomyces cerevisiae/S. mikatae, and Schizosaccharomyces japonicus being the four main yeast species detected (Wang et al., 2019). Yeast diversity and dynamics associated with the spontaneous fermentation of Vidal blanc ice-wine were also studied. The data showed that non-Saccharomyces yeasts were predominant in the initial stages, while Saccharomyces yeasts dominated in the final stages of fermentation as evinced by traditional culture-dependent and high-throughput sequencing methods (Li et al., 2018). Up to the present, there have been no reports on the study of yeast resources of ‘Beihong’.
In this study, the dynamic changes of wild yeast population during the spontaneous fermentations of ‘Beihong’ from Guiyang region were analyzed using high-throughput sequencing technology and culture-dependent approaches, which may provide theoretical support for the development and utilization of these native yeast resources.
Spontaneous fermentation and sample collection Berries of ‘Beihong’ were picked in september 2019 from three different vineyards in Kaiyang town, Guiyang city of Guizhou, China. The ‘Beihong’ grape was randomly selected from different picking sites in the same vineyard with the five parallel repetitions, and kept in sterile ice box, and transported to the laboratory. All the berries of ‘Beihong’ obtained from the three different vineyards were mixed to get the homogeneous sample and divided into three paralleled group. Healthy and undamaged grape berries were selected and pressed to obtain its must. Spontaneous laboratory fermentation was conducted with 600 mL of grape must in 1 000 mL sterile flasks at 28 °C in constant conditions in triplicate. Samples were prepared at 0, 1, 3, 5, 7, and 15 days (designated as H0, H1, H3, H5, H7 and H15) during the fermentation. Each sample was prepared and divided into two parts, one part used for traditional microbial separation, and the other one used for DNA extraction and high-throughput sequencing (Fig. 1).

Scheme of yeast diversity analysis of ‘Beihong’ during spontaneous fermentation using high-throughput sequencing and culture-dependent approaches
Yeast isolation and identification Samples were serially diluted and cultured on Wallerstein laboratory nutrient agar (Haibo, Qingdao, China) in triplicate at 28 °C for 5 days (Lorenzini et al. 2018). The yeast colonies on each plate were counted and calculated after incubation. For each plate, 30 colonies were randomly selected and isolated as representative yeast populations. Pure isolates on the Wallerstein laboratory nutrient agar were photographed and classified according to their morphotype. The representative yeasts were further subjected to molecular identification.
Genomic DNA was extracted using column yeast DNA purification kit (B518257, Sangon biotech, Shanghai, China) according to the manufacturer's instructions. DNA was quantified and then used as template for 26S rDNA D1/D2 domain sequences amplification with the primers NL1 (5′-GCATATCAATAAGCGGAGGAAAAG-3′) and NL4 (5′-GGTCCGTGTTTCAAGACGG-3′). Thermal cycles were as follows: 95 °C for 5 min; followed by 35 cycles at 94 °C for 30 s, 55 °C for 30 s, 72 °C for 50 s; and a final extension at 72 °C for 10 min. Electrophoresis of 1% agarose gel stained with GeneGreen, a kind of fluorochrome that could bind and displays the DNA molecules, was performed to analyze the quality of PCR products. Positive products were sent to Sangon Biotech Co., Ltd (Shanghai, China) for purification and sequencing. The sequences of the D1/D2 domain of 26S rDNA were aligned by BLAST (basic local alignment search Tooli).
Illumina high-throughput sequencing The total DNA was extracted directly from each sample by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China) using E.Z.N.A. soil DNA Kit (Omega Bio-tek; Norcross, GA, USA). The DNA concentration and purification were measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, USA). PCR amplification were performed using primers SSU0817F (5′-TTAGCATGGAATAATRRAATAGGA-3′) and 1196R (5′-TCTGGACCTGGTGAGTTTCC-3′) for amplifying V5 to V7 region of 18S rDNA, and purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA). Purified and pooled amplification libraries were paired-end sequenced (2 × 300) on the Illumina MiSeq platform (Illumina, San Diego, USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China).
Raw sequence reads were demultiplexed, quality-filtered, merged and clustered into OTUs with a 97% similarity cutoff using UPARSEii), and chimeric sequences were identified and removed using UCHIME (Edgar et al., 2011). The taxonomy of the acquired OTUs was analyzed using the RDP Classifier Bayesian algorithmiii) against the SILVA databaseiv) with a confidence threshold of 70%.
Basic agronomic trait and must chemical composition of ‘Beihong’ The color of ‘Beihong’ berry from Guiyang region was dark blue (Fig. 2), and the average weight of berry was 1.55 g. The grape must was red, no fragrance, and had a pH of 3.39 and a sugar content of approximately 18.2 °Brix.

External morphological characteristics of grape berries of ‘Beihong’
Fungal diversity determined by high-throughput sequencing approach
Coverage could evaluate the cover degree of operational taxonomic uints (OTUs) OTUs with low abundance in the sample (Nilsson et al., 2019; Staniszewska et al., 2019), and in this study the coverage of all the spontaneous fermentation samples was 1.00, indicating that the OTUs coverage of low abundance in this study was fully, and this sequencing could represent the real situation of the microbial population. A total of 281 009 932 sequencing bases, 735 802 effective sequences with an average length of 381.90 nt and 167 OTUs were obtained from eighteen samples at the 97% similarity level.
In high-throughput sequencing, species diversity of samples can be comprehensively assessed by shannon, simpson, sobs, ace and chao1 indexs. Generally, the larger of shannon index and the smaller the simpson index suggesting the higher species diversity in samples, and conversely, the lower it is (Reuter et al., 2015; Rogers et al., 2016). As shown in Table 1, the shannon indexes of sample H0 and H1 were larger, while the simpson indexes were smaller, indicating richer species diversity at the beginning of fermentation (H0) and the first day of fermentation (H1) of spontaneous fermentation. On the contrary, in the middle (H7) and late stage (H15) of spontaneous fermentation, shannon index and simpson index became smaller, indicating fewer species in these samples. Moreover, the sobs, ace and chao 1 indexes also applied to further analyze species diversity during spontaneous fermentation. Sobs, ace and chao 1 indexes of H0 were the highest, followed by H1, H3, H5, H7 and H15, that was consistent with the analysis results of shannon and simpson indexes. In briefly, these results indicated that with the continuous spontaneous fermentation of ‘Beihong’, the species diversity and richness gradually decreased.
| Samples | Shannon index | Simpson index | Sobs index | Ace index | Chao1 index |
|---|---|---|---|---|---|
| H0 | 2.04 | 0.25 | 47.00 | 56.52 | 49.98 |
| H1 | 2.13 | 0.18 | 42.33 | 44.95 | 43.53 |
| H3 | 0.83 | 0.52 | 22.33 | 28.30 | 24.67 |
| H5 | 0.17 | 0.93 | 13.67 | 16.03 | 15.17 |
| H7 | 0.02 | 1.00 | 7.67 | 11.69 | 8.67 |
| H15 | 0.07 | 1.00 | 4.33 | 2.00 | 4.33 |
A total of 143 genera and 147 fungus species were obtained from spontaneous fermentation of ‘Beihong’ samples. The sequences of top 50 species accounted for 99.99% of the total sequences. The top 50 fungal species was clustered in heatmap and shown in Fig. 3. The red color indicates the high species abundance in the sample, while the green color means the low species abundance. Samples of H0 and H1 were clustered in the same branch, indicating the similar species composition of these two samples. The middle and late fermentation stages (H3, H5, H7 and H15) were clustered in another branch. Pathogen was the major species mycete of the top 50 fungal species. There were only 11 yeast species including Hanseniaspora uvarum, Candida boidinii, Pichia terricola, Candida agrestis, Golubevia pallescens, Sporidiobolus pararoseus, Vishniacozyma peneaus, Starmerella bacillaris, Schizosaccharomyces japonicus, Clavispora Candida and Saccharomycetaceae were detected in spontaneous fermentation of ‘Beihong’ samples.
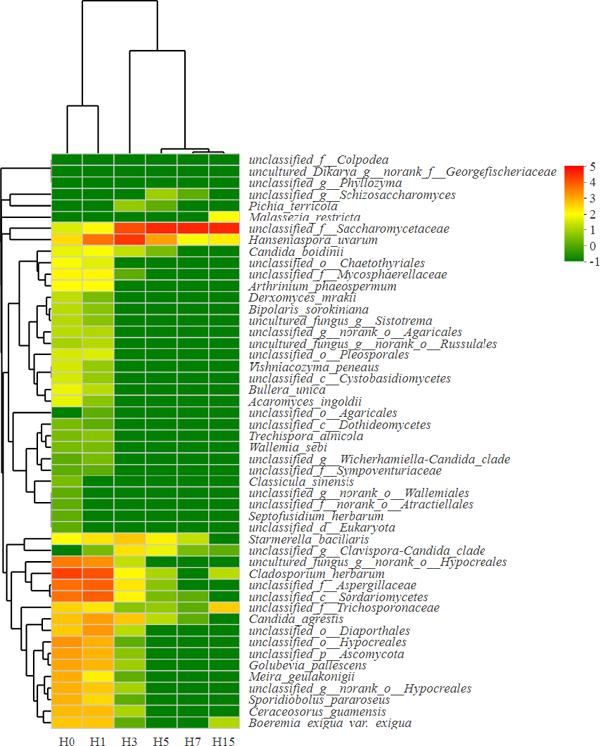
Heatmap and dendrogram of top 50 fungal species in spontaneous fermentation of ‘Beihong’
Identification of yeast isolates by culture-dependent approach
A total of 180 isolates were obtained from the spontaneous fermentation process. As shown in Fig. 4, 12 different yeast species were observed by culture-dependent approaches according to the morphological characteristics of their colonies on Wallerstein laboratory nutrient agar. The colors and morphologies of the colonies found in this study were summarized in Table 2.

The photographs of yeast colony morphotypes on the Wallerstein laboratory nutrient agar isolated from spontaneous fermentation of ‘Beihong’
| Number | Colony color | Colony topography |
|---|---|---|
| A | Yellowish white | Knoblike, Convex, smooth and opaque surface |
| B | Dark red | Convex, wrinkled surface |
| C | Slightly yellow to cream | Convex, wrinkled and opaque, irregular border |
| D | white | Convex, smooth and opaque surface |
| E | Cream, Blue green in the center | Convex, smooth and opaque surface, edge of flat |
| F | Dark red | Knoblike, Convex, smooth and opaque surface |
| G | Cream to white at the edge | Convex, smooth and opaque surface, edge of flat |
| H | White, Yellowish green in the center | Knoblike, Convex, smooth and opaque surface |
| I | White, center and edge dark green | Convex, smooth and opaque surface |
| J | White to dark green in the center | Convex, smooth and opaque surface |
| K | Dark green | Convex, smooth and opaque surface |
| L | Yellowish green | Convex, smooth and opaque surface |
Molecular methods were conducted to further confirm these isolates by comparing 26S rDNA D1/D2 domain sequences in Genbank. Results demonstrated that the yeast isolates species mainly from Ascomycota and Basidiomycota, belonged to 9 genera including Saccharomyces, Sporobolomyces, Pichia, Wickerhamomyces, Candida, Rhodosporidiobolus, Zygosaccharomyces, Starmerella and Hanseniaspora. At species level, the yeast species found were Saccharomyces cerevisiae, Sporidiobolus pararosenus, Pichia kluyveri, Wickerhamomyces anomalus, Candida oleophila, Rhodosporidiobolus ruineniae, Meyerozyma guilliermondii, Zygosaccharomyces bailii, Starmerella bacillaris, Hanseniaspora opuntiae, Hanseniaspora uvarum and Hanseniaspora clermontiae (Table 3).
| Isolate No. | Picture No. | Morphotype | D1/ D2 domain of 26S rDNA sequence/(identity %)/accession number |
|---|---|---|---|
| 1 | A | Saccharomyces cerevisiae | Saccharomyces cerevisiae/99.83%/ HM191639.1 |
| 2 | B | - | Sporidiobolus pararosenus/ 99.65%/NG_067316.1 |
| 3 | C | Pichia sp. | Pichia kluyveri/99.82%/HM123740.1 |
| 4 | D | - | Wickerhamomyces anomalus/100.00%/ MG773345.1 |
| 5 | E | Candida sp. | Candida oleophila/99.82%/KC510069.1 |
| 6 | F | Rhodosporidiobolus sp. | Rhodosporidiobolus ruineniae/99.82%/ LC178827.1 |
| 7 | G | - | Meyerozyma guilliermondii/100.00%/ KY952840.1 |
| 8 | H | Zygosaccharomyces bailii | Zygosaccharomyces bailii/99.82%/ EU441913.1 |
| 9 | I | - | Starmerella bacillaris/99.8%/J880968.1 |
| 10 | J | Hanseniaspora sp. | Hanseniaspora opuntiae/99.82%/ MN371931.1 |
| 11 | K | Hanseniaspora uvarum | Hanseniaspora uvarum/99.82% MN371949.1 |
| 12 | L | Hanseniaspora sp. | Hanseniaspora clermontiae/99.83%/ KC510056.1 |
Dynamic changes of yeast community composition during the fermentation by high-throughput sequencing and culture-dependent approaches
The dynamic changes of yeast flora in the spontaneous fermentation process of ‘Beihong’ were shown in Fig. 5, most variety of microorganism species existed at the beginning of fermentation (H0) and the first day of fermentation (H1), and the variety of microorganisms rapidly decrease as the fermentation continues (H3 and H5), and at the middle stage of fermentation most microorganisms died and disappeared. H. Uvarum is a kind of yeast commonly found in various grape varieties, which mainly existed in the early stage of spontaneous fermentation, and increased rapidly on the first day (H1), reached its maximum value on the third day (H3), then decreases sharply, and maintained at a very low level until the end of fermentation. The abundance of unclassfied_f_ Saccharomycetacease was very low at the beginning of fermentation, and increased dramatically from the third day of fermentation (H3), and then maintained very high throughout the fermentation progress (H5, H7, H15).

Histograms with the percent of community abundance on species level during the spontaneous fermentation of ‘Beihong’
In addition, phylogenetic tree was constructed by MEGA-X software based on neighbor joining method to further explore the species information of unclassfied_f_ Saccharomycetacease dominating from the fifth day until the fermentation finished (Kumar et al. 2018). It was shown that unclassfied_f_ Saccharomycetacease with the closest evolutionary relationships with S. cerevisiae, which clustered in the same branch (Fig. 6). Therefore, it was deduced that most non-saccharomyces yeast participated in fermentation process in the early stage of fermentation, and S. cerevisiae played the predominant roles from the middle stage until the late stage of spontaneous fermentation process of ‘Beihong’.

Phylogenetic tree of yeast strains detected by high-throughput sequencing method constructed by MEGA-X software based on neighbor joining method
Culture-dependent approach was also carried out to verify the yeast population dynamic changes, and results indicated that most yeast species existed at the initial stage of fermentation with a low abundance, and rapidly died at the second or the third day of spontaneous fermentation (Table 4). S. cerevisiae was present at relatively low abundance in samples H0 and H1, increased from the third day (H3), and maintained dominant species from H5 to H15. Hanseniaspora. sp. was found at the initial period, got its maximum at the third day, and then sharply decreased (Table 4).
| Species | Yeast community composition (%) | |||||
|---|---|---|---|---|---|---|
| H0 | H1 | H3 | H5 | H7 | H15 | |
| Saccharomyces cerevisiae | 3.33% | 16.67% | 30% | 90% | 90% | 100% |
| Sporidiobolus pararosenus | 10% | 10% | 0% | 0% | 0% | 0% |
| Wickerhamomyces anomalus | 6.67% | 10% | 6.67% | 6.67% | 10% | 0% |
| Candida oleophila | 10% | 10% | 0% | 0% | 0% | 0% |
| Rhodosporidiobolus ruineniae | 16.67% | 3.33% | 0% | 0% | 0% | 0% |
| Hanseniaspora. sp. | 6.67% | 13.33% | 60% | 3.33% | 0% | 0% |
| Meyerozyma guilliermondii | 16.67% | 16.67% | 3.33% | 0% | 0% | 0% |
| Pichia kluyveri | 13.33% | 10% | 0% | 0% | 0% | 0% |
| Zygosaccharomyces | 13.33% | 6.67% | 0% | 0% | 0% | 0% |
| Starmerella bacillaris | 3.33% | 3.33% | 0% | 0% | 0% | 0% |
As an eurasian Vitis hybrid cultivar with a superior cold and disease resistances, ‘Beihong’ had been widespread in many wine regions and selected for wine making in China. However, studies on the fungal diversity this cultivar was scarce. Compared to other grape variety such as Vitis vinifera L., Tempranillo and Vitis davidii Föex (Raymond et al., 2017; Hierro et al., 2006), ‘Beihong’ was less explored. It was well known that grape variety was a main factor in microbial terroir (Li et al., 2010). Therefore, this study was the first to use high-throughput sequencing and culture-dependent technologies to comprehensively analyze and monitor the indigenous yeast diversity and theirs dynamic changes during the spontaneous fermentation of ‘Beihong’ from Guiyang region. Our results demonstrated that 11 and 12 different yeast species were detected via high-throughput sequencing and culture-dependent approaches, respectively (Fig. 2 and Fig 3). H. uvarum, S.pararosenus and S. bacillaris were the yeast species detected by both high-throughput sequencing and culture-dependent approaches. But, some yeast species, such as C. boidinii, P.terricola, C. agrestis and G. pallescens only found in high-throughput sequencing results, and S. cerevisiae, P.kluyveri, W.anomalus, C.oleophila, R.ruineniae, M.guilliermondii, Z.bailii, H.opuntiae and H.clermontiae were the species specially observed by culture-dependent approach.
High-throughput sequencing technique is a powerful approach with the advantages of high speed, simple operation, comprehensive information and high efficiency (Soon et al., 2013), and had been widely used to investigate microbial diversity in various of samples such as human gut (Pallen et al., 2010), fermented food (Zhao et al., 2016), and grapes must (Wei et al., 2018), etc. However, high-throughput sequencing maybe lead to the preferential amplification of shorter sequences when using ITS primers for the target region analysis, because of the uneven ITS region lengths among fungi (Wang et al., 2019). In this study, V5 to V7 region of 18S rDNA instead of ITS region was amplified. However, some ‘uncultured’ and ‘unclassified’ species were also obtained when using high-throughput sequencing approach (Fig.3). In addition, S. cerevisiae, usually as the dominant yeast species during the late stages of spontaneous fermentations, just identified as unclassfied_f_ Saccharomycetacease (Fig. 3). This phenomenon may be attributed to the insufficient sequencing depth and limited coverage of the comparison database for the yeast diversity investigation. Indeed, the databases used for fungal diversity analysis did not include S.cerevisiae. Therefore, both high-throughput sequencing and culture-dependent technologies were indispensable for the investigation yeast diversity and dynamics in a comprehensive manner.
Generally, non-Saccharomyces yeast mainly presented in the early stage of fermentation because they were sensitive to ethanol, and Saccharomyces yeast successively dominant the middle stage and the late stage of fermentation (Jolly et al., 2003; Wang et al., 2016; Contreras et al., 2015). In the present study, most non-Saccharomyces yeast also detected in the initial stage of spontaneous fermentation of ‘Beihong’, and S. cerevisiae got predominate from the fifth day until the spontaneous fermentation finished (Fig.4). In oenology, the role of non-Saccharomyces yeast has been reevaluated, the utilization of mixed starter cultures composed by Saccharomyces and non-Saccharomyces yeasts is an approach of growing importance for winemakers to improve wine aroma complexity (Padilla et al., 2016; Liu et al., 2016). It was reported that H. uvarum contributed to increasing the wine organoleptic quality and to simultaneously reduce the volatile acidity (Tristezza et al., 2016). Here, we pointed out that Hanseniaspora sp. including H. opuntiae, H. uvarum and H. clermontiae were the dominant species at the third day of spontaneous fermentation (Fig. 4, Table 4). However, fewer descriptions of H. opuntiae and H. clermontiae were reported on the quality of fermented wines. Evidences implied that W. anomalus could adapt to a broad range of growth conditions in terms of osmolarity, temperature, and pH range values, and was a good producer of acetate esters, fruity acetate esters, and other volatiles, that exerted a positive effect on wine aroma (Padilla et al., 2018). W. anomalus was also observed in the early stage of spontaneous fermentation by culture-dependent technology (Table 4). Therefore, the oenological properties of these non-Saccharomyces yeasts identified and isolated in this research should be checked to further promote the selection of species conferring typical wine traits.
In summary, traditional microbial cultivation method and high-throughput sequencing approach were performed to investigate the yeast community and theirs dynamic changes during the spontaneous fermentation of ‘Beihong’. The result demonstrated that the composition of yeast species revealed by the two methods was quite different. Moreover, non-Saccharomyces yeasts were predominant in the initial stages of fermentation and then rapidly decreased, while Saccharomyces yeast sharply increased during the process. Hanseniaspora sp. and S. cerevisiae were the main yeast isolates at the third day of fermentation. Taken together, the present study was a first step in exploring untapped yeast resources of ‘Beihong’ of Guiyang region, showing the basic information on indigenous yeast diversity, which was helpful to screen these native yeasts for producing wines with regional characteristics.
Acknowledgements This work were supported by the Science and Technology Program of Guizhou Province (Talents of Guizhou Science Cooperation Platform [2017]5789, [2018]5603), Innovation Group Research Project from Guizhou Provincial Education Department (KY 2017046), and High-level Talent Research Funding Project of Guizhou Institute of Technology (XJGC20190625).
Conflict of interest There are no conflicts of interest to declare.