原著論文
トルコギキョウ[Eustoma grandiflorum (Raf.) Shinn.]由来のフラボノイド 3′- 水酸化酵素遺伝子(EgF3′H)の単離及び F3′H 機能欠失変異アサガオ[Ipomoea nil (L.) Roth.]を用いた EgF3′H の異種発現による相補性検定
2015 年 84 巻 2 号 p. 131-139

詳細
2015 年 84 巻 2 号 p. 131-139
A full-length cDNA of a putative flavonoid 3'-hydroxylase (F3'H) gene encoding a key enzyme in the production of cyanidin was cloned from a lisianthus (Eustoma grandiflorum) petal. Lisianthus F3'H (EgF3'H) shares 75.1, 73.8, and 68.2% amino acid identity with Arabidopsis thaliana, Ipomoea nil, and Petunia hybrida, respectively. RT-PCR revealed that wild-type lisianthus flowers accumulated higher levels of F3'H mRNA during the early stages of development than in the late stages. The accumulated F3'H transcript levels in leaves were similar to those in flowers in the early stages of development. Overexpression of lisianthus F3'H cDNA altered flower color from red to blue in the I. nil cultivar ‘Violet’, which lacks a functional F3'H gene. In addition, the transgenic ‘Violet’ plants accumulated cyanidin and peonidin at similar levels to wild-type I. nil. Taking these findings together, this study demonstrates that EgF3'H functions as a flavonoid 3'-hydroxylase with a role in the synthesis of cyanidin and peonidin pigments.
Flower color is the most outstanding, important trait of floricultural plants. Flavonoids are important plant pigments that are responsible for producing many floral colors in the visible spectrum. The most abundant and predominant flavonoid pigments are the anthocyanins, which produce a wide range of colors, such as orange, pink, red, magenta, purple, violet, and blue (Schwinn and Davies, 2004). The anthocyanin biosynthetic pathway is well established, and many genes encoding related enzymes have been cloned (Holton and Cornish, 1995). The degree of B-ring hydroxylation has the greatest effect on anthocyanin-based flower color. Hydroxylation of the B-ring is regulated by the activities of two cytochrome P450 enzymes (CytP450s), flavonoid 3'-hydroxylase (F3'H) and flavonoid 3',5'-hydroxylase (F3'5'H). The cDNAs of the F3'H and F3'5'H genes were first isolated from petunia (Petunia hybrida) by Brugliera et al. (1999) and Holton et al. (1993), respectively.
Lisianthus, or prairie gentian [Eustoma grandiflorum (Raf.) Shinn.], a member of the family Gentianaceae, is native to the central and southern United States (Shinners, 1957). This species is one of the most popular cut flowers in Japan due to its large flower, long stem, and extended vase life and ranked eleventh in the Dutch cut-flower market in 1995 (Ledger et al., 1997). The wild-type lisianthus plant bears a purple flower whose color can be attributed to the presence of delphinidin-derived pigments. Cultivars with flower colors such as white, cream, pink, red, magenta, and purple have been generated by commercial breeding, particularly in Japan (Ledger et al., 1997).
In lisianthus, two types of full-length F3'5'H cDNA clones have been isolated from the petals of purple flowers (Nielsen and Podivinsky, 1997; Noda et al., 2004; Shimada et al., 1999); however, the molecular and biochemical characterization of lisianthus F3'H has not been described.
Japanese morning glory [Ipomoea nil (L.) Roth.] (Pharbitis nil Choisy) is a traditional horticultural plant in Japan from which a number of spontaneous mutants with various flower colors have arisen, particularly since the late Edo era (1806–1860). Most of these mutant lines have been maintained, and many of their mutations have been genetically defined (Iida et al., 1999, 2004; Imai, 1927). Nearly all of the structural and regulatory genes involved in anthocyanin biosynthesis have been identified (Chopra et al., 2006; Morita et al., 2006). Consequently, I. nil is considered a valuable experimental system for flower pigment studies, along with Petunia (Ishikawa et al., 2002). However, I. nil also has several traits that are disadvantageous to its use as a model for molecular genetic studies. For example, it is very recalcitrant to the regeneration of plantlets from undifferentiated cultured cells and tissues and thus is very difficult to transform genetically (Bapat and Rao, 1977; Sangwan and Norreel, 1975). This problem has recently been overcome, however, by the development of more efficient and reliable transformation methods (Kikuchi et al., 2005; Ono et al., 2000). Furthermore, mutant strains, EST clones, linkage maps, and transgenic lines have been developed, collected and distributed by the National BioResource Project (http://www.nbrp.jp/localeAction.do?lang=en).
In the present study, we successfully isolated the F3'H cDNA from lisianthus flowers and confirmed its function as a flavonoid 3'-hydroxylase that functions in cyanidin synthesis by transgenic complementation of the heterologous plant species I. nil cultivar ‘Violet’, which has a magenta mutation that eliminates F3'H activity (Hoshino et al., 2003).
Three cultivars of lisianthus, ‘Mellow Lavender’ (mauve-flowered), ‘Royal Violet’ (violet-flowered), and ‘Asuka no Kurenai’ (red-flowered), were used in this study. The seeds of ‘Mellow Lavender’ and ‘Royal Violet’ were purchased from Takii Seed Co., Ltd. (Kyoto, Japan), and ‘Asuka no Kurenai’ was purchased from Sakata Seed Co., Ltd. (Yokohama, Japan). The progeny obtained by self-pollination of these cultivars were designated as ME, RV, and AK, respectively (Shimizu et al., 2011). Lisianthus plants were grown for gene cloning and RT-PCR analysis was performed as described previously (Hashimoto et al., 2004; Shimizu et al., 2011; Uddin et al., 2002). RNA was extracted from leaves as well as petals at six different floral developmental stages: 1) bud less than 20 mm long; 2) bud between 20 mm and 30 mm; 3) bud more than 30 mm long and tightly closed; 4) bud more than 30 mm long and loosely closed; 5) opening flower; and 6) fully opened flower. Plant materials were immediately frozen in liquid nitrogen and stored at −80°C prior to RNA extraction or high-performance liquid chromatography (HPLC) analysis.
The seeds of I. nil cultivar ‘Violet’ were purchased from Marutane Trading Co., Ltd. (Kyoto, Japan), and those of the wild-type strain ‘TKS’ (Tokyo kokei standard) were provided by Dr. A. Hoshino (National Institute for Basic Biology). The cell suspension culture was prepared from immature embryos of I. nil and used for Agrobacterium-mediated transformation, according to the method described by Shimizu et al. (2003).
Characterization of anthocyanidin compositionThe anthocyanidin compositions of lisianthus and Japanese morning glory were analyzed by HPLC using previously described procedures (Hashimoto et al., 2002, 2004; Uddin et al., 2001, 2002), with some modifications. Flower petals were harvested at the fully opened stage and stored at −80°C until use. For anthocyanidin analysis, frozen petal samples were macerated in 6 mL of 1% HCl methanolic solution and allowed to equilibrate overnight at 4°C. After filtering, to obtain the aglycones of anthocyanins, 2 mL of filtrate was hydrolyzed with 4 mL of 2 N HCl at 100°C for 2 h in a heating block. The hydrolyzed extracts were refiltered (0.45 μm), and the filtrate was analyzed by HPLC.
Filtered extracts were analyzed on a TSKgel ODS-80Ts QA Column (4.6 mm i.d. × 150 mm; TOSOH Co., Tokyo, Japan). The column was attached to a Jasco HPLC system (Jusco Co., Tokyo, Japan) equipped with a gradient pump (PU-2089), mixer (MX-2082-32), autosampler (AS-2051), diode-array PDA HPLC detector (MD-2010), and column oven (CO-2060). For analytical-scale HPLC, the mobile phase consisted of solvent A (1.5% phosphoric acid) and solvent B (1.5% phosphoric acid, 20% formic acid, and 25% acetonitrile). The mobile phase composition was controlled by the HPLC system software and varied from 70% solvent A and 30% solvent B to 30% solvent A and 70% solvent B over 35 min. The flow rate was 0.8 mL·min−1, the injection volume was 10 μL and UV detection was performed at 525 nm. The total area under the peaks at 525 nm was quantified by referring to calibration curves of anthocyanidin standards.
Cloning and sequencing of cDNA encoding flavonoid 3'-hydroxylase (EgF3'H)Degenerate oligonucleotide primers (Forward-A: TTY AAY ATH GGI GAY TTY ATH CC; Forward-B: TTY ACI GCI GGI ACI GAY AC; Reverse-C: GGR TCY CKI CCD ATI GCC CA; Reverse-D: RCA DAT YCT TCK ICC IGC ICC) were designed based on the highly conserved amino acid regions of plant F3'H genes. Total RNA was extracted from 1 g (fresh weight, FW) of stage 3–4 petals derived from ME, exhibiting mauve flower color with delphinidin and cyanidin as the major anthocyanidins, using the RNeasy Maxi Kit (QIAGEN GmbH, Venlo, Netherlands), and poly(A)+ RNA was purified using Oligotex-dT30 Super (TAKARA BIO INC., Otsu, Japan). cDNA was synthesized using the GeneRacer Kit (Invitrogen, Carlsbad, CA, USA) and used as a template for PCR amplification using the primers listed above. The PCR reaction mixture (10 μL) consisted of 1× PCR buffer for KOD -Plus- Ver. 2 (TOYOBO, Osaka, Japan), 0.2 mM each dNTP, 1.5 mM MgSO4, 0.2 units of KOD -Plus- DNA polymerase, 1 μM forward primer and reverse primer, and 1 μL of cDNA as the template. The thermal cycling conditions were 94°C for 2 min; followed by 30 cycles of 94°C for 30 s, 52°C for 30 s, and 68°C for 60 s; and a final elongation step at 68°C for 10 min. The PCR products were separated by agarose gel electrophoresis and purified using a QIAEX II Gel Extraction Kit (QIAGEN GmbH). Purified fragments were sub-cloned into a pCR-Blunt-II-TOPO® vector (Invitrogen) for sequencing.
To obtain the full-length sequence of the F3'H cDNA clone, the cDNA templates derived from lisianthus petals were further subjected to PCR amplification with KOD -Plus- DNA polymerase (TOYOBO) using the following gene-specific primers: Eg3'HRev (GCC GAA TAC CCT CCT TCC AAG C) or Eg3'HFor (GCA TCC TCA CGC GAT CTA TAG C) and GeneRacer 5' primer (CGA CTG GAG CAC GAG GAC ACT G) or 3' primer (GCT GTC AAC GAT ACG CTA CGT AAC G). The PCR reaction mixture (10 μL) consisted of 1× PCR buffer for KOD -Plus- Ver. 2 (TOYOBO), 0.2 mM each dNTP, 1.5 mM MgSO4, 0.2 units of KOD -Plus- DNA polymerase, 1 μM forward primer and reverse primer, and 1 μL of cDNA as a template. The thermal cycling conditions were 94°C for 2 min; followed by 5 cycles of 94°C for 30 s and 72°C for 60 s; 5 cycles of 94°C for 30 s, 70°C for 30 s, and 72°C for 60 s; 20 cycles of 94°C for 30 s, 68°C for 30 s, and 72°C for 60 s; and a final elongation step at 72°C for 10 min. The resulting PCR product was concentrated by ethanol precipitation and resuspended in 10 μL of TE for nested PCR. For nested PCR, the following sets of gene-specific primers were used: Eg3'HRev-Nest (GTC TAA GAA CGC ATC GAA ACG C) or Eg3'HFor-Nest (CAC AAA TGC TCT GGC GAG AGT G) and GeneRacer 5' Nested Primer (GGA CAC TGA CAT GGA CTG AAG GAG TA) or 3' Nested Primer (CGC TAC GTA ACG GCA TGA CAG TG). The reaction mixture for the nested PCR was the same as that for the first PCR mixture, and the thermal cycling conditions were 94°C for 2 min; followed by 30 cycles of 94°C for 30 s, 58°C for 30 s, and 68°C for 60 s; and a final step at 68°C for 10 min. The PCR products were separated by agarose gel electrophoresis and subjected to sequence analysis. Nucleotide sequences were determined using an ABI PRISM® 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The obtained sequence was used in a homology search using the BlastX program (Ref) against the nucleotide data collected in the DDBJ database (http://www.ddbj.nig.ac.jp/). Other analyses were primarily conducted using the GENETYX program (GENETYX Co., Tokyo, Japan). A phylogenetic tree of the deduced amino acid sequence was generated using the CLUSTAL W (Thompson et al., 1994) and TREEVIEW programs (Page, 1996).
Reverse transcription-PCR (RT-PCR)Total RNA was extracted from 100 mg (FW) of lisianthus petals using the Plant Total RNA Extraction Kit Mini (Viogene, New Taipei City, Taiwan). To eliminate residual DNA, the RNA samples were treated with DNase I (amplification grade) (Invitrogen). DNase I was inactivated by the addition of 1 μL of 25 mM EDTA solution to the reaction mixture, followed by heating at 65°C for 10 min. The first-strand cDNA was synthesized from DNase I-treated RNA using the PrimeScript II 1st strand cDNA Synthesis Kit (TAKARA BIO INC.). The amount of RNA for each cDNA synthesis reaction mixture was adjusted to 1 μg per reaction. The PCR reaction mixture (10 μL) consisted of 1× Ex Taq Buffer, 0.2 mM each dNTP, 0.25 units of Ex TaqTM HS (TAKARA BIO INC.), 0.5 μM forward primer (ATG GAA ATG ATC AAT GAT TCT CCC) and reverse primer (TCA TTT AAC ATA CAT AGA GCT AGC AAG), and approximately 50 ng of first-strand cDNA as the template. The thermal cycling conditions were 95°C for 5 min; followed by 30–35 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 1 min; and a final elongation step at 72°C for 5 min. Lisianthus ubiquitin (GenBank accession number AB049409) (Oka et al., 2001) was also amplified by RT-PCR as an internal control using specific forward (EgUbi-F, 5'-ATC CAG TGA CAC CAT CGA CA-3') and reverse primers (EgUbi-R, 5'-TGT TGT AGT CGG CGA GAG TG-3'). The RT-PCR products were subjected to agarose gel electrophoresis. Fluorescent images of the gel were captured after staining with GelRed (Biotium, Inc., Heyward, CA, USA).
Heterologous expression of the EgF3'H gene in I. nilThe coding regions of the F3'H cDNAs were amplified by PCR using a gene-specific forward primer containing a start codon, EgF3'HcozATG (CAA ATG GAA ATG ATC AAT GAT TCT CCC) (first methionine codon is underlined), and a reverse primer containing a deduced stop codon, EgF3'HRv (TCA TTT AAC ATA CAT AGA GCT AGC AAG) (stop codon is underlined). The PCR reaction mixture (10 μL) consisted of 1× PCR buffer for KOD -Plus- Ver. 2, 0.2 mM each dNTP, 1.5 mM MgSO4, 0.2 units of KOD -Plus- DNA polymerase (TOYOBO), 1 μM forward primer and reverse primer, and 0.1 μL of cDNA as the template. The thermal cycling conditions were 94°C for 2 min; followed by 30 cycles of 94°C for 30 s, 60°C for 30 s, and 68°C for 90 s; and a final elongation step at 68°C for 10 min. The amplified fragments were cloned into GATEWAY vector pCR8/GW/TOPO according to the pCR8/GW/TOPO TA cloning kit manual (Invitrogen), and then the coding region of the F3'H gene was re-cloned into pH2GW, a binary vector for Agrobacterium tumefaciens-mediated transformation (Karimi et al., 2002) using Gateway BP and LR Clonase Enzyme Mix (Invitrogen). The binary vector contains two antibiotic resistance genes for plant (hygromycin) and bacterial (spectinomycin) selection. The resulting recombinant plasmid was designated pGWEgF3'H and contained the coding region of F3'H downstream of the CaMV 35S promoter and upstream of the 35S terminator. This chimeric gene was named “35S::EgF3'HCR”.
Agrobacterium-mediated transformation of I. nil was conducted as previously described with some modifications (Kikuchi et al., 2005; Ono et al., 2000; Otani and Shimada, 1998; Shimizu et al., 2003; Yoneda and Nakamura, 1987). Agrobacterium tumefaciens AGL0 cells carrying recombinant pGWEgF3'H were cultured overnight at 28°C in 5–10 mL of liquid LB medium (10 g·L−1 tryptone, 5 g·L−1 yeast extract, and 10 g·L−1 NaCl) supplemented with 100 mg·L−1 spectinomycin. The bacterial cells were harvested by centrifugation and resuspended in MS medium with 10 g·L−1 sucrose (pH 5.8), and the concentration was adjusted to attain final absorbance of 0.2–2.0 at 600 nm (OD600). Cell suspension cultures of I. nil ‘Violet’ were mixed with the A. tumefaciens suspension, and the calli were then transferred to MS medium supplemented with 200 μM acetosyringone, 10 g·L−1 sucrose, and 3.2 g·L−1 gellan gum (pH 5.8) for co-culture. After three days, cell suspension cultures were transferred to EI medium (Jia and Chua, 1992) supplemented with 50 mg·L−1 hygromycin, 1 tablet·L−1 Augmentin Combination Tablets 250RS (GlaxoSmithKline K.K., Tokyo, Japan), 60 g·L−1 sucrose, and 3.2 g·L−1 gellan gum to select transformed calli. After three to four weeks of culture, hygromycin-resistant (Hmr) embryoids were formed on the calli. The Hmr embryoids were transferred to the same fresh medium and cultured for three to four more weeks for proliferation and maturation. Then, the embryoids were transferred to EMG medium (Jia and Chua, 1992) supplemented with 1 tablet·L−1 Augmentin Combination Tablets 250RS for shoot formation. Root formation and growth of transgenic plants were conducted according to the methods described by Shimizu et al. (2003). Expression of the transgenic F3'H gene was confirmed by RT-PCR using the EgF3'cozATG and EgF3'HRv primers. The gene for 18S ribosomal RNA (rRNA) was also amplified by RT-PCR as an internal control using the TaqMan® primers for Eukaryotic 18S rRNA (Applied Biosystems).
A partial cDNA fragment of EgF3'H was amplified by PCR using degenerate primers designed based on common motifs of plant F3'H genes, and its full-length cDNA was then cloned by RACE PCR with primers designed from the previously amplified fragment. Nucleotide sequence analysis revealed that the EgF3'H cDNA (GenBank accession number AB922856) was 1757 bp long and contained a putative open reading frame (1560 bp) encoding 519 amino acid residues. Multiple alignments revealed a high degree of amino acid sequence identity among the lisianthus F3'H (EgF3'H) and three reported F3'H sequences (Fig. 1); however, the first 35 amino acid residues and the region between residues 263 and 274 varied among these F3'Hs. The deduced amino acid sequences of EgF3'H share 384/511 (75.1%), 347/509 (68.2%), and 378/512 (73.8%) identical residues with those of P. hybrida (Brugliera et al., 1999) (AF155332), A. thaliana (Schoenbohm et al., 2000) (AL133421), and I. nil (Hoshino et al., 2003) (AB113261), respectively. A phylogenetic tree of plant F3'H and F3'5'H proteins is shown in Figure 2. As expected, EgF3'H and F3'H from Gentiana triflora (Nakatsuka et al., 2005) (DQ218417) belong to the same subgroup, most likely because both species belong to Gentianaceae. In addition, this subgroup is closer in evolutionary distance to other plant F3'Hs and further from the plant F3'5'H group.

Multiple alignment of the deduced amino acid sequences of F3'H from lisianthus (AB922856; EgF3'H), Ipomoea nil (AB113261; InF3'H), Petunia hybrida (AF155332; PhF3'H), and Arabidopsis thaliana (AL133421; AtF3'H). Identical amino acid residues are indicated in black shaded boxes. The dots indicate the conserved proline-rich residues that are crucial for P450 topology. The underlines indicate the P450 motifs AGTDTS and FGAGRRICAG and the F3'H-specific motifs GGEK (see details in the text).
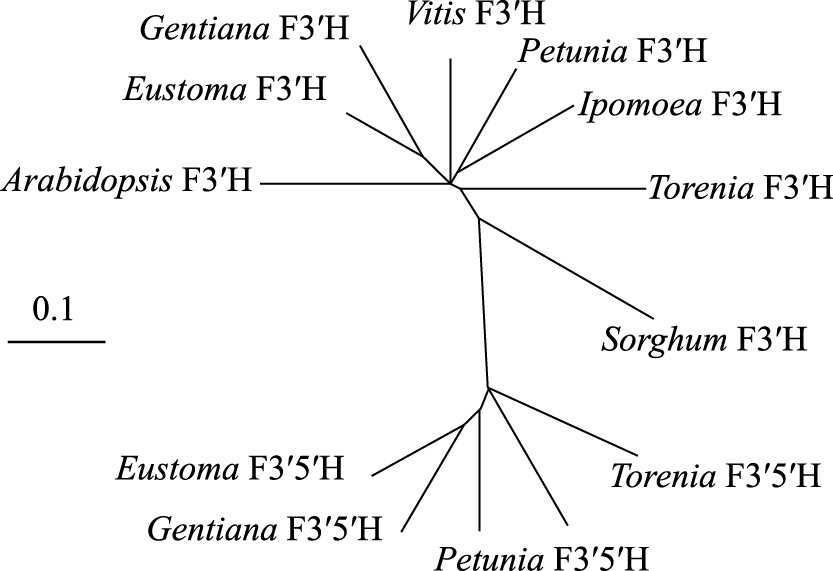
A molecular phylogenetic tree of the deduced amino acid sequences of EgF3'H and those of known plant F3'H and F3'5'H genes. The accession numbers in the GenBank/EMBL/DDBJ databases are as follows: Eustoma F3'H (E. grandiflorum, AB922856), Vitis F3'H (Vitis vinifera, AJ880357), Petunia F3'H (P. hybrida, AF155332), Ipomoea F3'H (I. nil, AB113261), Gentiana F3'H (Gentiana triflora, DQ218417), Arabidopsis F3'H (A. thaliana, AL133421), Torenia F3'H (Torenia hybrida, AB057672), Sorghum F3'H (Sorghum bicolor, AY675075), Petunia F3'5'H (P. hybrida, Z22544), Torenia F3'5'H (T. hybrida, AB012925), Eustoma F3'5'H (E. grandiflorum, AB078957), and Gentiana F3'5'H (G. triflora, D85184).
Sequence analysis revealed that the EgF3'H protein contains several domains conserved in plant F3'Hs. EgF3'H has a proline-rich motif (Fig. 1, dotted sequences) that is crucial for the formation of the correct conformation of microsomal P-450 molecules (Murakami et al., 1994; Yamazaki et al., 1993). The carboxy-terminal region contains a conserved “FX2GXRXCXG” (447–456) motif; the cysteine residue in this motif is involved in binding the heme iron (Werck-Reichhart et al., 2002) (Fig. 1, underlined sequence). EgF3'H also bears the conserved motif “AGTDTS” (311–316), which reportedly forms an oxygen-binding pocket required for its catalytic activity (Chapple, 1998) (Fig. 1, underlined sequence). In addition, EgF3'H has a “GGEK” (429–432) motif that is conserved in all identified F3'H genes (Brugliera et al., 1999) (Fig. 1, underlined sequence). Thus, multiple sequence alignment and the phylogenetic tree strongly suggest that the EgF3'H cDNA clone isolated from flower petals of E. grandiflorum encodes a flavonoid 3' hydroxylase enzyme.
Analysis of EgF3'H gene expression by RT-PCRIn this study, we analyzed EgF3'H gene expression in three lisianthus lines with different anthocyanidin compositions: one exhibiting a violet flower color with delphinidin as the major anthocyanidin type; one exhibiting a mauve flower color with delphinidin and cyanidin as the major anthocyanidins; and one exhibiting a red flower color with pelargonidin as the major anthocyanidin. The expression of EgF3'H was investigated in lisianthus leaves and petals at six different stages of floral development (Fig. 3A). The RT-PCR profiles revealed relatively high expression levels of the 1.6 kb transcript of EgF3'H at early stages of flower development in the three lisianthus lines analyzed. EgF3'H expression tended to decrease in all lines when the flowers reached stages 4–6, in which the corolla was fully pigmented and open (Fig. 3B). High-level expression of EgF3'H was also observed in the leaf tissue of all lines tested.

Flower development and EgF3'H expression in the petals of three lisianthus lines: ME, AK, and RV. (A) Developmental stages of the flower and leaf. (B) RT-PCR analysis of EgF3'H expression in the petals of six developmental stages of the flower as well as leaves.
Wild-type I. nil strain ‘TKS’ has blue flowers, while I. nil ‘Violet’ has reddish flowers due to a mutation in the F3'H gene (Fig. 4A) (Hoshino et al., 2003). We utilized this genetic trait of the latter strain to determine whether the cloned EgF3'H was active; specifically, the 35S::EgF3'HCR chimeric gene was introduced into I. nil ‘Violet’ by Agrobacterium-mediated transformation.

Alteration of flower color due to heterologous expression of EgF3'H in transgenic plants of I. nil ‘Violet’. (A) Photographs (overhead view) of flowers from ‘TKS’ (left, a wild-type I. nil that expresses functional F3'H), ‘Violet’ (middle, an I. nil plant harboring mutated F3'H) and “35S::EgF3'HCR” (right, a transgenic ‘Violet’ line carrying EgF3'H). (B) HPLC profiles of anthocyanidins accumulated in the petals of ‘TKS’ (left), non-transgenic ‘Violet’ (middle), and a representative transgenic ‘Violet’ plant expressing EgF3'H (right). Cy, cyanidin; Pg, pelargonidin; Pn, peonidin. (C) Photographs of petioles from non-transgenic parental I. nil ‘Violet’ (left) and a representative transgenic ‘Violet’ expressing EgF3'H (right).
Eight plants were regenerated from independent calli and designated No. 1–8. These independent regenerated plants were derived from approximately 0.4 mL of suspension calli infected with A. tumefaciens AGL0 harboring pGWEgF3'H. Six regenerated plants (No. 1–5 and 7 in Table 1) exhibited apparent phenotypic alterations. As shown in Figure 4A and C, non-transgenic I. nil ‘Violet’ exhibited reddish flowers and red petioles, whereas the six transgenic plants bore blue flowers and black petioles, as in wild-type I. nil ‘TKS’ (Fig. 4A, C). The other two plants (No. 6 and 8 in Table 1) did not exhibit flower color changes but bore black petioles.
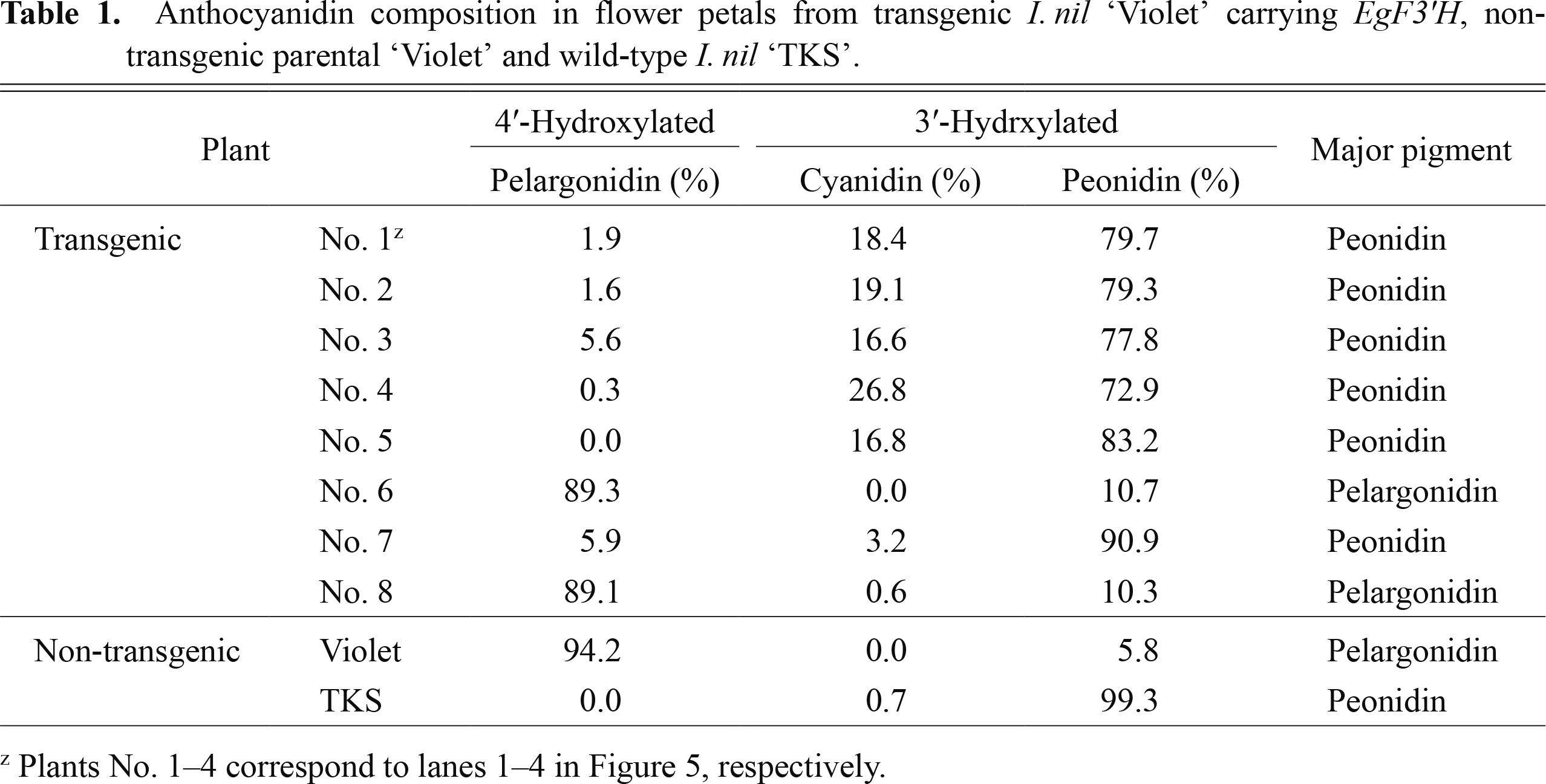
Anthocyanidin composition in flower petals from transgenic I. nil ‘Violet’ carrying EgF3'H, non-transgenic parental ‘Violet’ and wild-type I. nil ‘TKS’.
Four of the eight regenerated plants (No. 1–4 in Table 1) were subjected to RT-PCR analysis to examine the expression of introduced EgF3'H, which revealed that all accumulated the mRNA of the introduced F3'H in their flower petals (Fig. 5), suggesting that the transgenes are active in these plants. HPLC profiles revealed a dramatic difference in anthocyanidin composition in the petals of non-transgenic and transgenic plants. 3'-Hydroxylated anthocyanidins (cyanidin and peonidin) accounted for 94.4–99.7% of the total anthocyanidin content in the flowers of the four transgenic plants, while a low occupancy (5.8%) of 3'-hydroxylated anthocyanidin (peonidin) was observed in non-transgenic ‘Violet’ plants (Fig. 4B; Table 1). These results strongly suggest that EgF3'H encodes a functional flavonoid 3'-hydroxylase that is involved in the synthesis of cyanidin and peonidin pigments.
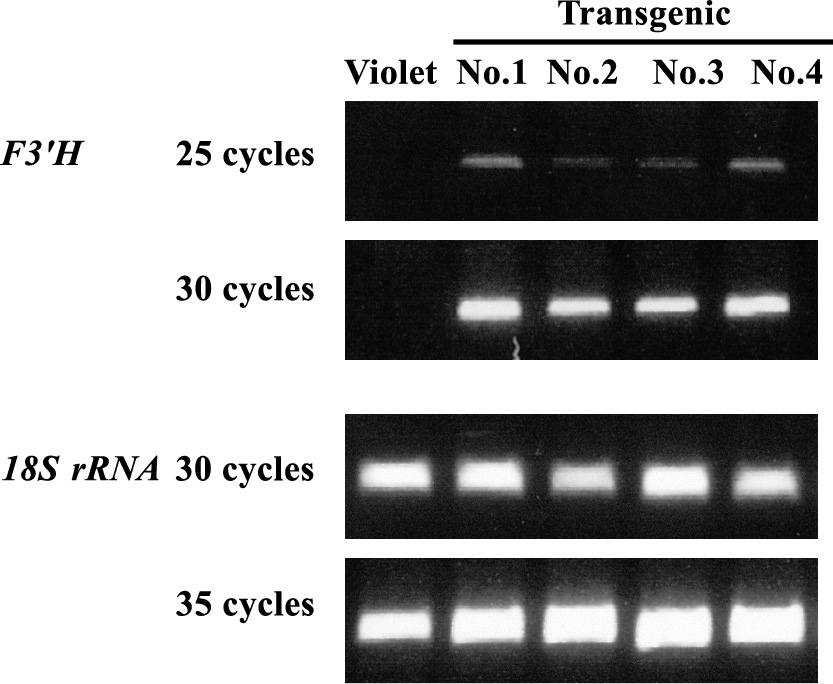
Semi-quantitative RT-PCR analysis of EgF3'H expression in petals from non-transgenic parental I. nil ‘Violet’ and four plants (No. 1–4) transformed with the corresponding gene. The lisianthus 18S rRNA gene was used as a loading control. The numbers on the left-hand side of the photographs indicate the numbers of PCR cycles.
In this study, we successfully cloned an F3'H homologue from lisianthus, EgF3'H. Multiple sequence alignment revealed that EgF3'H shares 75.1%, 73.8%, and 68.2% amino acid identity with the F3'H genes of P. hybrida, I. nil, and A. thaliana, respectively. EgF3'H contains several motifs conserved in plant P450s: proline-rich residues, the heme domain (FGAGRRICAG), and the binding pocket motif (AGTDTS) (Chapple, 1998; Xu et al., 2007). In addition, EgF3'H contains an F3'H-specific motif (GGEK) (Brugliera et al., 1999).
Nakatsuka et al. (2005) classified the genes encoding enzymes involved in flavonoid biosynthesis in Gentiana into three groups. Group I includes CHS (chalcone synthase) and CHI (chalcone isomerase), which are expressed in all stages of flower development. Group II includes F3'H and FSII (flavone synthase II), which are expressed in early stages of flower development. Group III includes F3H (flavanone 3-hydroxylase), F3'5'H, DFR (dihydroflavonol 4-reductase), ANS (anthocyanidin synthase), 3GT (anthocyanin 3-O-glucosyltransferase), and 5AT (anthocyanin 5-aromatic acyltransferase), which are all expressed in the late developmental stages. Davies et al. (1993) observed that PAL (phenylalanine ammonia-lyase), CHS, and CHI are expressed at all stages of lisianthus flower development, while DFR is expressed only at the late stages. Noda et al. (2004) also reported that lisianthus CHS, CHI, and F3H were coordinately expressed during all stages of floral development, whereas F3'5'H, ANS, and DFR were expressed in late stages and FLS (flavonol synthase) was expressed in early stages. In this study, EgF3'H gene expression was observed in early stages of flowering (Fig. 3B). This expression pattern in floral organs is similar to that of FLS in lisianthus (Noda et al., 2004) and F3'H and FSII in Gentiana (Nakatsuka et al., 2005). Taken together, these results suggest that EgF3'H is expressed during the early stages of flower development in lisianthus, similar to FLS. Expression patterns similar to that of lisianthus F3'H have been observed in other plant species, including Petunia (Holton et al., 1993), Torenia (Ueyama et al., 2002), Glycine max (Zabala and Vodkin, 2003), and G. triflora (Nakatsuka et al., 2005).
The EgF3'H gene was also found to be expressed at a high level in lisianthus leaf tissue (Fig. 3B), allowing us to speculate that anthocyanin synthesis is in a preparatory state in lisianthus leaves, similar to that observed in early-stage floral organs. Arabidopsis and Perilla express F3'H in their leaves, and this expression is likely associated with anthocyanin accumulation in the corresponding organs (Kitada et al., 2001; Schoenbohm et al., 2000). By contrast, EgF3'H expression in lisianthus leaves does not appear to be associated with anthocyanin accumulation because other flavonoid biosynthesis genes, such as CHS, CHI, and even FLS, are not abundantly expressed in the leaves (Noda et al., 2004). Taken together, these results suggest that, in leaves, EgF3'H has function(s) other than anthocyanin biosynthesis that remains to be explored. For instance, F3'H is supposed to be involved not only in anthocyanin biosynthesis but also in flavone biosynthesis (Kitada et al., 2001).
Nielsen and Podivinsky (1997) and Noda et al. (2004) did not detect the F3'5'H gene transcript in pink flower lisianthus lines that lack delphinidin but accumulate pelargonidin. Thus, by analogy with F3'5'H, we anticipated marked variation in F3'H expression among the three lisianthus lines with different anthocyanidin compositions; however, such marked variation of F3'H expression was rarely detected among these lines in our RT-PCR analysis (Fig. 3). Although slight differences in F3'H expression levels were observed in early stages of flowering, the temporal expression patterns were consistent in these strains. There are several potential explanations for our results. First, regulatory mechanisms at the protein level, such as post-translational processing or enzyme activity control of the F3'H protein, could explain the observed differences in anthocyanin accumulation among the lines, despite the nearly equal mRNA levels. Second, slight differences in F3'H mRNA levels observed in the early stages of flower development might determine the anthocyanin compositions of the different plant lines. Additional reasons, such as the presence of a mutated allele of F3'H that encodes an enzyme with low activity levels in the lisianthus plant line RV with high levels of delphinidin, should also not be ruled out.
Genetic transformation approaches have been applied for functional analyses of cloned genes or cDNAs, such as the regulatory and enzyme-encoding genes involved in flavonoid biosynthesis. In these studies, both homologous and heterologous plants were used as hosts for genetic transformation. For example, Brugliera et al. (1999) first cloned a F3'H homologue from petunia and confirmed its function by transforming homologous plants with F3'H containing a mutation. Ueyama et al. (2002) produced torenia plants overexpressing their own F3'H and confirmed that this gene can alter flower color in transgenic plants. Other groups have used heterologous hosts for the same purpose. Shimada et al. (1999) reported the heterologous expression in Nicotiana tabacum of two F3'5'H genes from P. hybrida and Eustoma russellianum (E. grandiflorum). Mori et al. (2004) reported heterologous expression in Petunia of the F3'5'H gene from Vinca major. Zufall and Rausher (2004) also confirmed the substrate specificity of the DFR enzyme and the function of the F3'H gene from I. quamoclit by transgenic experimentation with heterologous Arabidopsis plants.
Similar to the studies described above, we utilized I. nil as a heterologous host for transgenic complementation of the F3'H gene cloned from lisianthus (Fig. 4) because an authentic magenta mutant of this species, I. nil ‘Violet’, has a nonsense point mutation in the F3'H gene (Hoshino et al., 2003). We successfully demonstrated that the lisianthus F3'H gene encodes a flavonoid 3'-hydroxylase that plays a role in anthocyanin production. In addition, our study further supports the use of I. nil as a very valuable model organism for molecular genetic studies involving flower pigments. This is the first report of both the cloning of the lisianthus F3'H gene and transgenic complementation using a heterologous host of Japanese morning glory carrying a mutant allele of the corresponding gene.
We are very grateful to Dr. A. Hoshino (National Institute for Basic Biology) for providing the I. nil strain ‘TKS’ seeds.