Review
Cardio-renal safety of non-steroidal anti-inflammatory drugs
2019 Volume 44 Issue 6 Pages 373-391
Details
2019 Volume 44 Issue 6 Pages 373-391
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most widely used therapeutic class in clinical medicine. These are sub-divided based on their selectivity for inhibition of cyclooxygenase (COX) isoforms (COX-1 and COX-2) into: (1) non-selective (ns-NSAIDs), and (2) selective NSAIDs (s-NSAIDs) with preferential inhibition of COX-2 isozyme. The safety and pathophysiology of NSAIDs on the renal and cardiovascular systems have continued to evolve over the years following short- and long-term treatment in both preclinical models and humans. This review summarizes major learnings on cardiac and renal complications associated with pharmaceutical inhibition of COX-1 and COX-2 with focus on preclinical to clinical translatability of cardio-renal data.
The conversion of arachidonic acid (AA) into prostaglandin H2 (PGH2) is catalyzed by cyclooxygenases (COXs). PGH2 is subsequently converted to other prostaglandins that regulate various physiologic functions (e.g., PGE2, PGI2, PGF2α, and thromboxane A2 [TXA2]) (Eling et al., 1990; Radi, 2009) (Fig. 1). COX-1 and COX-2 are two membrane-anchored isoenzymes. While COX-1 is constitutively expressed under physiologic conditions, COX-2 is induced under conditions of inflammation and injury (Parente and Perretti, 2003; Radi, 2009). In the cardio-renal systems, AA metabolites and PGs generated by COXs play a role in maintaining homeostasis such as renal blood flow, electrolyte balance, and platelet functions (Fitzgerald, 2004; Khan et al., 2013; Radi, 2009; Sellers et al., 2010).
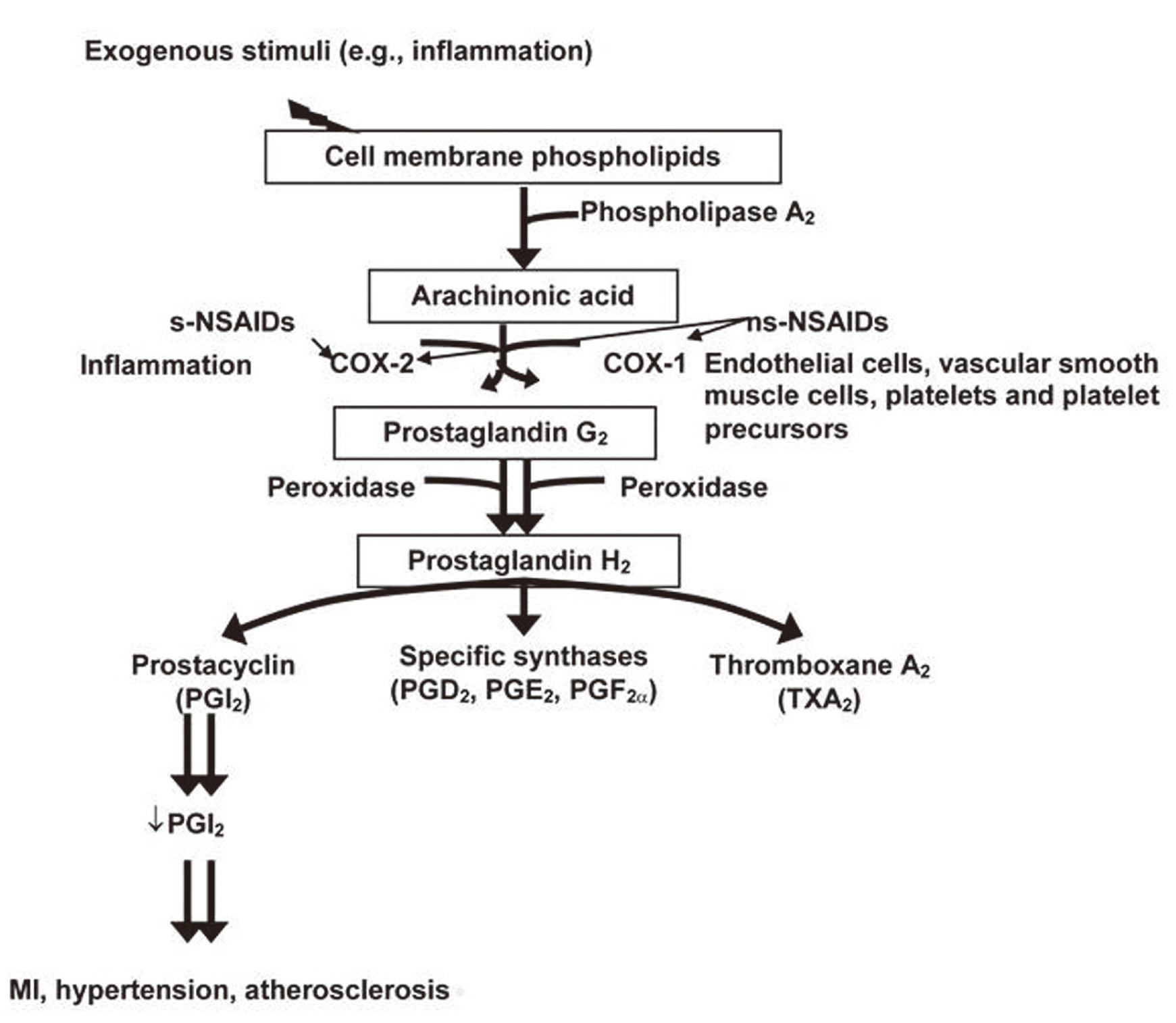
Prostaglandins (PGs) pathophysiologic role in cardio-renal system safety under normal and pathological conditions and the effects of pharmaceutical drugs such as nonselective and selective nonsteroidal anti-inflammatory drugs (ns-NSAIDs and s-NSAIDs) on these systems. Initially, arachidonic acid is liberated from cell membrane phospholipid by phospholipase A2 when there is exogenous inflammatory stimulus. Subsequently, the two cyclooxygenase isoforms, COX-1 and COX-2, facilitate the conversion of arachidonic acid into PGs. COX-1 isoform is expressed by the cardio-renal system cellular components (e.g., endothelial cells, platelets, vascular smooth muscle cell, and kidney interstitial cells), whereas there is an upregulation of COX-2 following inflammatory insult and under pathological conditions (e.g., myocardial infarction [MI], atherosclerosis, chronic kidney disease, and glomerulonephritis). Non-NSAIDs (e.g., carprofen, ketoprofen, indomethacin, and phenylbutazone) inhibit COX-1 and COX-2; s-NSAIDs (e.g., celecoxib, lumiracoxib, robenacoxib, and rofecoxib) spare COX-1 and inhibit only COX-2. Published In: Sellers RS, Radi ZA, Khan NK. Pathophysiology of cyclooxygenases in cardiovascular homeostasis, Veterinary Pathology 47(4):601-13. Copyright © 2010 SAGE Publications. DOI: 10.1177/0300985810364389.
There are two main inhibitors of COX activity: (1) non-selective non-steroidal anti-inflammatory drugs (ns-NSAIDs), and (2) selective NSAIDs (s-NSAIDs). The ns-NSAIDs (e.g., naproxen, ibuprofen, diclofenac) inhibit both COX-1 and COX-2 at pharmacologic doses, whereas s-NSAIDs (e.g., celecoxib, lumiracoxib, robenacoxib) inhibit more selectively COX-2 to minimize COX-1-mediated toxicities (e.g., gastrointestinal (GI) and kidney injury) (Khan et al., 2013; Parente and Perretti, 2003; Radi, 2009; Radi and Khan, 2006). Adverse cardiovascular (CV) outcomes with some selective COX-2 inhibitors (Rofecoxib) led to its withdrawal from the market in 2004. Preclinical studies could not recapitulate these CV events in standard toxicity studies and experimental laboratory models. The potential for cardio-renal drug safety events associated with COX inhibition has been thoroughly investigated in the last few years, including a large randomized, multicenter, double-blind, non-inferiority trial involving patients who were at increased cardiovascular risk and had rheumatoid arthritis or osteoarthritis treated with a s-NSAID (celecoxib) and two ns-NSAIDs (ibuprofen and naproxen) (Nissen et al., 2016; Ruschitzka et al., 2017; Solomon et al., 2017; Krotz et al., 2005). In this review, we summarize the role of COXs in cardio-renal homeostasis, examine the comparative pathophysiological differences in COX inhibition across species, and share learnings from cardio-renal pharmaceutical drug safety profiles in preclinical species and humans.
The cardio-renal expression of COX-1 and COX-2 in normal tissues in humans and laboratory animals are summarized below and in Table 1. Under normal conditions in heart of humans and animals, COX-1 is expressed in endothelial cells and vascular smooth muscle (Koki et al., 2002; Stanfield et al., 2001; Zidar et al., 2007, 2009; Radi, 2009; Sellers et al., 2010) (Table 1). In normal human heart, COX-1 is expressed in endothelial and smooth muscle cells of blood vessels and in endothelial cells of the endocardium (Zidar et al., 2007). In rats, COX-1 is expressed in endothelial cells of the heart, but not in cardiomyocytes (Sellers et al., 2010). In heart tissue of dogs, COX-1 expression was noted in fibrous connective tissue of the tricuspid valve and chordae tendinae (Stanfield et al., 2001). COX isoform expression under static or shear stress was investigated using porcine aortic endothelial cell (PAEC) culture (Potter et al., 2011). PAEC had detectable levels of COX-1 with low levels of COX-2, and shear stress did not affect the relative expression of COX-1 and COX-2 at either 24 hr or 7 days. Endothelial cell medium contained low levels of the prostacyclin metabolite 6-keto-PGF1α when cells were grown under static or sheared conditions for up to 7 days (Potter et al., 2011). In another study, bovine aortic endothelial cells (BAEC) exposed to high shear stress showed increased COX-2 expression up to 5 hr (Russell-Puleri et al., 2017). Increased COX-1 expression has been noted in the aortic endothelial cells from hypertensive rats (Kim et al., 2001; Tang and Vanhoutte, 2008). Aging experiments in Wistar-Kyoto rats showed overexpression of COX-1, COX-2, TXA synthase, PGD synthase, mPGE synthase-2, and PGF synthase in endothelial cells, and COX-1 and PGE2 receptors in smooth muscle cells (SMC) (Tang and Vanhoutte, 2008). Furthermore, in aging and senescence, endothelium has greater propensity to release COX-derived vasoconstrictive PGs (Silva et al., 2017). The impact of COX inhibition in aged humans and animals might have a greater significance clinically (Sellers et al., 2010). Experimental studies of the aortic endothelium from aged pigs and rats demonstrated increased COX-1 and/or COX-2 expression (Kim et al., 2001; Tang and Vanhoutte, 2008). It has been suggested that endothelial senescence is associated with reduced anti-platelet activity and promotes a prothrombotic response (Silva et al., 2017). In humans with mean age of 66 ± 3 years with coronary artery disease, improved coronary artery endothelium-dependent vasodilation was noted with COX-2 inhibition (Chenevard et al., 2003).
| Renal | Human | Monkey | Dog | Rat |
|---|---|---|---|---|
| COX-1 | + | + | + | + |
| Collecting ducts | ||||
| Papillary Interstitial cells | ++ | ++ | +++ | +++ |
| Renal Vasculature | + | + | +/- | + |
| COX-2 | +/- | +/- | +/- | - |
| Glomerulus | ||||
| TAL | - | - | + | + |
| Macula densa | +/- | - | + | + |
| Collecting ducts | - | - | - | - |
| Papillary Interstitial cells | - | - | + | + |
| Renal Vasculature | +/- | +/- | +/- | - |
| Cardiac | ||||
| COX-1 | - | - | - | - |
| Cardiomyocyts | ||||
| Endothelial cells | ++ | ++ | ++ | ++ |
| Vascular smooth muscle cells | ++ | ++ | ++ | ++ |
| COX-2 | ||||
| Cardiomyocytes, endothelial cells, vascular smooth muscle cells | - | - | - | - |
Expression included the following grading system: (-) = no expression, (+) = minimal expression; (++) = moderate expression; (+++) = strong expression. TAL = Thick Ascending Limb.
In contrast to COX-1, COX-2 is generally not expressed in normal endothelial or vascular smooth muscle cells; however, it is rapidly induced under conditions of vascular trauma or inflammation (Sellers et al., 2010; Dannenberg et al., 2001; Koki et al., 2002; Smith et al., 2000; Zidar et al., 2007) (Table 1). In normal coronary arteries of humans (Baker et al., 1999) and in non-atheromatous aorta of humans (Schönbeck et al., 1999), limited COX-2 expression has been reported. COX-2 expression in the heart is limited and restricted to aortic vascular endothelial cells in the dog and rat, and no expression has been identified in human aorta (Kawka et al., 2007). In cardiomyocytes of normal humans, dogs, and rodents, COX-2 expression was only rarely evident (Zidar et al., 2007). In heart tissue of mice and rats, scant COX-2 and high COX-1 expression were noted (Testa et al., 2007). In cultured neonatal rat normal cardiomyocytes, treatment with sodium salicylate significantly increased COX-2 expression and induced prostaglandin D2 release (Ock et al., 2018).
Genetically engineered mice can be used to understand the role of PGs in cardiac physiology. However, because COX-2 null mice have significantly underdeveloped kidneys and poor renal function, distinguishing whether the cardiac change of myocardial fibrosis is primary or secondary is challenging (Sellers et al., 2010). Genetically engineered mice with reduced COX-2 expression levels (i.e., knock-down) are generally interpreted to be similar to that which would be induced by s-NSAIDs had no changes in heart rate, or blood pressure (Seta et al., 2009). Thus, COX-2 is essential for normal cardiac physiology, but reductions in COX-2 expression (versus complete deletion of COX-2 expression) do not significantly impact myocardial function (Sellers et al., 2010). Over-expression of COX-2 in the heart of mice showed no differences in cardiac function, weight, or morphologic changes by histologic examination under normal conditions (Inserte et al., 2009).
To maintain a balance between vasoconstriction, vasodilation and anti- and pro-thrombotic events, opposing effects of PGs on cardiac homeostasis and vascular tone are noted. Such balance is mediated via PGI2 and platelet-derived TXA2, and to a lesser extent PGE2. TXA2, primarily COX-1-dependent, is mainly platelet-derived and is the predominant AA metabolite in platelets (Rossi et al., 1996). Only COX-1 was expressed in platelets in both humans and preclinical toxicology species and these cells do not have an inducible COX-2 isoform (Kay-Mugford et al., 2000; Zidar et al., 2009). TXA2 causes local vasoconstriction, platelet aggregation, and smooth muscle cell proliferation. For example, genetic depletion of TXA2/thromboxane prostanoid (TP) receptor signaling in mice prevented microvascular dysfunction (Chiang et al., 2018). Mice over-expressing the TXA2-receptor have increased neo-intimal hyperplasia, intimal cell proliferation, and platelet activation after mechanical artery injury (Cheng et al., 2002). Under normal physiologic conditions, reductions in COX-2 did not notably alter blood pressure in mice (Seta et al., 2009). Thus, increased platelet aggregation was likely attributable to COX-2 related changes in vascular wall physiology (Sellers et al., 2010). PGI2-receptor deficient mice with vascular injury are more prone to thrombotic events compared with WT mice (Cheng et al., 2002; Murata et al., 1997). Thus, PGI2 is essential for balancing the pro-thrombotic and vasoconstrictive effects of TXA2 (Fitzgerald, 2004; Kearney et al., 2004). While PGE2 can cause vasodilation and can modulate the effects of TXA2 on platelet aggregation, it does not directly participate in homeostasis. Interestingly, while there is a balance between TXA2 and PGI2, experimental studies in PGI2 receptor-deficient and TXA2 over-expressing mice have demonstrated lack of increased platelet aggregation or thrombotic events under physiological conditions (Cheng et al., 2002; Murata et al., 1997). Therefore, other homeostatic mechanisms and molecules (e.g., nitric oxide [cardioprotective and reduces thrombosis and atherosclerosis], carbon monoxide) might play a role in thrombotic cascades under normal conditions (Marcus et al., 2002; Lundberg et al., 2015).
In the kidney, COX-1 is the most abundant isoform localized in the renal vasculature, collecting ducts, and papillary interstitial cells across various species (Radi and Ostroski, 2007; Khan et al., 1998, 2013; Radi, 2009) (Table 1). The ontogeny of renal COX expression has been investigated in a study in developing rats where kidney COX expression changes were examined at various ages (Ogawa et al., 2001). COX-1 expression did not change with age in either the cortex or medulla. COX-2 expression was highest in 1-week-old rats and lowest in 4- and 8-week-old rats. In another study of adult normal human kidney, COX-1 expression was noted in the collecting duct cells, interstitial cells, endothelial cells, and smooth muscle cells of glomerular vasculature. In Madin Darby Canine Kidney (MDCK) cells, no COX-1 expression was detected (Pelletier and Padhi, 2015). In young dogs, no COX-1 expression was noted in diseased or normal kidneys (Yabuki et al., 2016). In fetal human kidney, COX-1 was primarily expressed in podocytes and collecting duct cells of the kidney. Expression levels of COX-1 in both cell types increased markedly from subcapsular to juxtamedullary cortex (Kömhoff et al., 1997). Another study examined COX-1 expression in normal human kidney tissues using autopsy samples. COX-1 was noted in blood vessels, interstitial cells, smooth muscle cells, and platelets (Zidar et al., 2009). No differences in COX-1 renal regional localization were noted among various species (Khan et al., 1998). Evidence across species (cow, dog, guinea pig, human, monkey, mouse, rabbit, rat, and sheep) demonstrated that high levels of COX-1 expression occurred in the collecting ducts (Câmpean et al., 2003; Khan et al., 1998, 2013) (Table 1). Low or high dietary salt did not affect COX-1 expression (Yang et al., 1998).
COX-2 is constitutively expressed in macula densa (MD) and thick ascending limb (TAL) of loop of Henle in the normal kidney and can be found in interstitial cells (Ferreri et al., 1999; Câmpean et al., 2003; Khan et al., 2013; Yang et al., 1999) (Table 1). COX-2 expression has been studied in human fetal kidneys ranging between 15 and 23 weeks of gestational age. Strong COX-2 expression was localized primarily in the MD and the TAL of the loop of Henle, and was rare in glomerular podocytes and vascular endothelial cells. A progressive decrease in COX-2 expression from the most immature nephrons adjacent to the metanephric regions to the well-developed nephrons in the middle to inner cortex was noted. In contrast to the adult human kidney, this temporal and spatial expression of COX-2 in the fetal kidney suggests that COX-2 enzyme may be involved in nephrogenesis (via cell differentiation and maintaining nephron renal blood flow to counterbalance the renin-angiotensin system), and its inhibition by NSAIDs during the third trimester may be responsible for fetal renal syndromes (Khan et al., 2001, 2013). Interestingly, salt supplementation partly reversed kidney morphological defects in COX-2-/- mice and improved kidney function (Slattery et al., 2017). The temporal expression of COX-2 throughout embryonic and fetal development was investigated in rats. COX-2 expression was not detectable in any tissues from developing embryos during gestation days 7 to 13, but was observed in the fetal growth period (gestation days 15 to 20) in several organs, including the kidney (Stanfield et al., 2003). Interspecies differences may exist for the involvement of COX-2 in the generation of renal PGs in states of normal and altered kidney function (Khan et al., 2013). For example, the normal rat and dog kidney have prominent constitutive COX-2 expression in the MD and thick ascending limb of loop of Henle; whereas COX-2 is absent at these sites in the normal nonhuman primate and human kidney (Sellers et al., 2004; Khan et al., 2013). In young dogs, low or undetectable COX-2 expression was noted in chronic kidney disease while normal kidneys had COX-2 expression in the MD (Yabuki et al., 2016). COX-2 levels also differ according to maturation, with high levels expressed in the MD and TAL of fetal kidneys and minimal expression upon renal maturation (Khan et al., 2013, 2001). COX-2 is expressed in rats MD and regulates tubuloglomerular feedback via two mechanisms: (1) generation of TxA2, and (2) reduction of neuronal nitric oxide synthase-dependent nitric oxide (Araujo and Welch, 2009). TXA2 receptors (TP) have been found in various regions of the kidney. Thromboxane synthase has been detected in the glomeruli of rat kidneys (Vitzthum et al., 2002). Injection of synthetic TxA2 into the renal artery of anesthetized pigs resulted in a dose-related decrease in renal blood flow (Cirino et al., 1990).
Various PGs, especially PGE2 and PGI2, are synthesized in the kidney, contributing to urine electrolyte balance via inhibition of the tubular transport function and regulation of renin-angiotensin-aldosterone system (RAAS) system activity. In fact, recent data showed that MD cell prorenin receptor activation results in stimulation of PGE2 release (Riquier-Brison et al., 2018). The MD-derived COX-2 products could counteract renal vasoconstriction in nitric oxide synthase inhibition-induced hypertension and maintain renin synthesis/secretion (Kommareddy et al., 2011). In uninephrectomized rats and mice, increased tensile stress and fluid flow shear stress increased COX-2 expression and stimulated PGE2 synthesis in podocytes (Srivastava et al., 2014). PGE2 can maintain GFR by dilating the afferent arterioles (Edwards, 1985; Schlondorff, 1993). In addition, medullary PGE2 promotes renal sodium excretion via the EP2 receptor. For example, mice placed on a high salt diet exhibited increased medullary COX-2 and microsomal prostaglandin E synthase-1 (mPGES1) expression (Chen et al., 2008). It is thought that renal medullary interstitial cell NFκB plays an important role in mediating renal medullary COX2 expression (He et al., 2014). In addition, COX-2 expression dramatically increased in the MD and cortical thick ascending limb, in both humans and rodents with salt and volume depletion (Harris et al., 1994; Kömhoff et al., 2000). Some human syndromes such as Bartter-like syndrome are associated with significant renal salt and water loss. In this syndrome, there is also marked increase in urine PGE2 excretion (Kömhoff et al., 2000). The MD cells synthesize and release PGE2 during reduced luminal salt content and this response is important in the control of renin release and renal vascular resistance during salt deprivation (Peti-Peterdi et al., 2003).
The pathophysiologic complications associated with modulation of COX-1 and/or COX-2 in cardio-renal systems are summarized below and in Table 2. Cyclooxygenase activity is induced in human atherosclerosis (Belton et al., 2003). Atherosclerosis is a complex inflammatory disease of medium and large sized vessels which results in intimal accumulation of lipids, macrophages, and other inflammatory infiltrates as well as smooth muscle cell proliferation leading to atherosclerotic plaques (Moubayed et al., 2007; Ross, 1993; Sellers et al., 2010). COX-1 expression was investigated in human atherosclerotic aortic plaques. COX-1 levels were only 1.1-fold higher in plaque tissue. There was no relationship between gender, age, or cause of death and COX-2 or COX-1 levels (McGeer et al., 2002). PGs promote atherosclerosis by altering the inflammatory response and expression of matrix metalloproteinases (MMPs) as well as by acting as a mitogen for cell types, including vascular smooth muscle cells (VSMCs). Up-regulation of MMP-2 and MMP-9 was noted in plaques and suggested, in part, to be mediated by COX-2 and COX-1 derived PGE2 (Cipollone and Fazia, 2006) and may contribute to plaque rupture (Sellers et al., 2010). In fact, administration of the s-NSAID, celecoxib, to rabbits with atherosclerosis demonstrated anti-atherosclerotic effects in aortic plaques via inhibition of COX-2 and MMP-9 by suppressing NF-κB activation (Li et al., 2018). TXA2 is clearly an important mediator of plaque development and progression (Cyrus et al., 2007). The source of TXA2 increase in atherosclerosis is primarily derived from COX-1 in platelets (Belton et al., 2003). Much of the PGI2 increases in atherosclerosis have been attributed to COX-2 up-regulation at the site of the plaque (Belton et al., 2000). In the region of atherosclerotic plaques, COX-2 expression is present in endothelial cells, VSMCs and macrophages (Belton et al., 2003). Mouse and rabbit models of atherosclerosis have COX-2 expression in fatty streaks and atherosclerotic vessels (Belton et al., 2000; Burleigh et al., 2005b; Hong et al., 2000; Stemme et al., 2000; Wong et al., 2001). These data indicate that in animal models of atherosclerosis, the expression of COX isoforms is comparable to that of humans (Sellers et al., 2010).
| Renal | Pathophysiologic Complications |
|---|---|
| ↑ COX-1 | Chronic kidney disease, hypertension models |
| ↑ COX-2 | Nephrogenic diabetes insipidus, chronic kidney disease, glomerulonephritis, passive Heymann nephritis, lupus nephritis, polycystic kidney disease, diabetic nephropathy, renal hypertension, unilateral ureteral obstruction, bilateral ureteral obstruction |
| Cardiac | |
| ↑ COX-1 | Atherosclerosis, myocardial infarction, myocardial ischemia/reperfusion |
| ↑ COX-2 | Atherosclerosis, myocardial infarction, myocardial ischemia/reperfusion, cardiomyocyte hypertrophy, hypertension, congestive heart failure |
↑ = Increased expression
The importance of PGs in the development of atherosclerosis has been evaluated in animal models. PGI2 has a protective role against the atherosclerosis, while TXA2 is important in the initiation and progression of atherosclerosis (Egan et al., 2005; Kobayashi et al., 2004). Modulation of platelet aggregation and leukocyte-endothelial cell interactions are contributing factors through changes in adhesion molecules, intercellular adhesion molecule-1 (ICAM-1) and platelet endothelial cell adhesion molecule-1 (PECAM-1). Some in vitro and mouse knockout data suggest that COX-2/PGI2 mechanotransduction is mediated by PECAM-1 (Russell-Puleri et al., 2017). Apolipoprotein E deficient (Apoe)-/-COX1-/- null mice fed a 1% cholesterol diet for 8 weeks demonstrated decreased atherosclerotic lesions (McClelland et al., 2009). These data implicate COX-1 in the early pathogenesis of atherosclerosis. Interestingly, platelet adhesion to vessel walls was not notably different between double mutant and controls under normal conditions; however, reductions in platelet adhesion were evident after vascular ligation injury in Apoe-/-COX1-/- mice. This suggests that COX-1 is also important in promoting platelet adhesion to damaged vessel walls and is likely mediated through TXA2 production (Sellers et al., 2010). Studies in hamsters have also demonstrated that PGI2 has atheroprotective properties and PGI2 analogs retarding atherogenesis is consistent with the genetically engineered mouse studies (Kowala et al., 1993).
Systemic blood pressure is primarily maintained by the RAAS system, and hypertension is often due to alterations in renal vascular homeostasis (Radi, 2009). PGE2, derived from both COX-1 and COX-2, promotes vasodilatation and, while not by itself thrombogenic, promotes the thrombogenic potential of TXA2 (Sellers et al., 2010; Gross et al., 2007). PGE2 was released from both ischemic and non-ischemic cardiac regions during myocardial ischemia in anesthetized dogs (Berger et al., 1976). Increases in PGI2 are the result of both COX isoforms in the endothelium (Belton et al., 2003). Mice lacking the receptor for PGI2 (IP KO) develop salt-sensitive hypertension, cardiac hypertrophy, and cardiac fibrosis. Deletion of the TxA2 receptor did not prevent the development of hypertension, but cardiac hypertrophy was ameliorated, and fibrosis was prevented in double knockouts of receptors for PGI2 and TxA2 (Francois et al., 2005). Decreased susceptibility to renovascular hypertension has been reported in mice lacking PGI2 receptor (Fujino et al., 2004). COX-1 null mice under conditions of reduced sodium intake have mild but significant reduction in blood pressure over control mice (Athirakul et al., 2001). Thus, COX-1 is important in maintaining blood pressure indirectly by maintaining sodium retention. In addition, blood pressure has a circadian pattern, with reductions in blood pressure during sleep (Kawada et al., 2005). COX-1 null mice have impaired reductions in blood pressure during sleep (Kawada et al., 2005), indicating that COX-1 may be important in maintaining normal circadian rhythms in blood pressure. COX-2 knock-down mice have no notable changes in systemic blood pressure as compared to control mice (Seta et al., 2009). These data indicate that, under normal conditions, COX effects on systemic blood pressure are primarily mediated through its actions on renal sodium retention/excretion (Sellers et al., 2010).
COX-2 is readily observed in damaged cardiac tissues both in humans and experimental animal models of myocardial disease (Sellers et al., 2010) (Table 2). In human hearts obtained after post-myocardial infarction (MI), COX-1 and COX-2 expression was present in inflammatory cells, myofibroblasts, and in capillaries in areas of granulation tissue and fibrosis. In human patients with heart failure and MI, abundant COX-2 expression has been detected in cardiomyocytes adjacent to infarcted regions as well as in inflammatory cells and vessels associated with regions of cardiac damage and scar formation (Abbate et al., 2004; Zidar et al., 2007). In addition, COX-2 expression has been identified in heart and peripheral blood of patients with septicemia and myocarditis (Zhang et al., 2018). In a rat model of chronic MI and heart failure, strong COX-2 expression was observed in the myocytes, endocardium, vascular endothelial cells, and macrophages in the viable tissue adjacent to the infarcted region, as compared to nominal expression noted in control rats (Saito et al., 2004). Mice with transient coronary artery occlusion followed by reperfusion and COX-2 over-expression in cardiomyocytes demonstrated improved contractility during the reperfusion phase and decreased infarction region compared with WT mice (Inserte et al., 2009). In both WT and COX-2 overexpressing mice, the PGE2, PGI2, and PGF2α increased, but not TXA2. However, only PGE2 was significantly higher in transgenic (TG) mice, suggesting that this molecule is important in the prevention of cell death during ischemia-reperfusion events. MI induced in microsomal prostaglandin E2 synthase-1 null mice revealed similar effects on post-MI survival, infarct size, and cardiac injury marker (cardiac troponin-I) levels as compared to control animals indicating that inflammation-induced PGE2 may not significantly contribute to the outcome in MI (Wu et al., 2009). Collectively, these data indicate that rodent models and human COX-2 responses in MI are similar (Sellers et al., 2010).
Over-expression of the PGE2 receptor gene in mice resulted in marked myocardial hypertrophy with increased interstitial fibrosis in the TG mice as compared with WT mice (Meyer-Kirchrath et al., 2009). These mice had marked increases in diastolic and systolic pressures at 5-6 weeks of age as compared to the WT littermates. In addition, the left ventricular ejection fraction was markedly decreased in the TG mice, suggesting reductions in contractility. Furthermore, COX-2 has been found to play a role in the hypertrophy of neonatal rat cardiomyocytes (Li et al., 2014). Thus, PGE2 and COX-2 might be factors in the development of myocardial hypertrophy.
In the renal system, COX-1 expression has been investigated in various pathological conditions (Table 2). Nephrogenic diabetes insipidus (NDI) is a renal disorder that is characterized by an inability to concentrate urine despite normal or elevated plasma concentrations of the antidiuretic hormone (ADH) arginine vasopressin (Morello and Bichet, 2001; Khan et al., 2012). In NDI, there is a peripheral resistance to ADH and polyuria (excessive or abnormally large production and/or passage of urine) with hyposthenuria (urine of low specific gravity), and polydipsia (excessive thirst) being the cardinal clinical manifestations of the disease (Khan et al., 2012). NSAIDs have been used to treat patients with NDI (Pattaragarn and Alon, 2003). Therefore, COX expression has been investigated in animal models of NDI. In a NDI rat model, a marked decrease in the expression of COX-1 in the inner medulla of the kidney and marginally reduced COX-1 expression in the inner stripe of outer medulla has been reported (Kotnik et al., 2005). In dogs and cats with chronic kidney disease (CKD), COX-1 expression was observed in the medullary distal tubules and collecting ducts, but no correlations with the severity of renal damage were detected (Yabuki et al., 2012). COX-1 expression was studied in two kidney-one clip (2K1C) Goldblatt hypertensive rat model. In this 2K1C hypertension model, the defect is highly dependent on the enhanced activity of the RAAS. COX-1 expression in the 2K1C model was localized to the extra-glomerular mesangium and distal convoluted tubules (DCT), connecting tubule, and cortical collecting duct except for the intercalated cells (Theilig et al., 2006). In a hypertension transgenic mouse model that express the human renin and angiotensinogen genes, the highest degree of renal COX-1 expression occurred in the DCT, cortical collecting ducts, and medullary collecting ducts, while mild-to-moderate COX-1 expression occurred in other microanatomic renal locations. Proximal convoluted tubules (PCT) lacked COX-1 expression (Radi and Ostroski, 2007).
COX-2 deficient mice exhibited severe disruption of renal development and function, suggesting an important role for COX-2 in renal development (Dinchuk et al., 1995; Morham et al., 1995; Khan et al., 2013). Enhanced COX-2 expression has been demonstrated in several renal pathologies such as glomerulonephritis (Schneider et al., 1999), passive Heymann nephritis (Heise et al., 1998), lupus nephritis (Tomasoni et al., 1998), CKD (Yabuki et al., 2012), hypertension, congestive heart failure, diabetic nephropathy, and obstructive nephropathy (Mohamed et al., 2013). In a streptozotocin-induced diabetes mouse model, increased podocyte expression of COX-2 increased susceptibility to development of diabetic nephropathy and the podocyte injury was due in part to increased expression and activity of the pro-renin receptor. These mice exhibited significant renal changes characterized by albuminuria, foot process effacement, and glomerular basement membrane thickening (Cheng et al., 2011). In a mouse model of autosomal dominant polycystic kidney disease (ADPKD), celecoxib treatment ameliorated ADPKD progression, and improved renal function (Monirujjaman and Aukema, 2019). COX-2 but not COX-1 expression was higher in kidney cystic epithelial cells and in the interstitium, and mPGES1 and PGI synthase levels were increased in a mouse model of PKD and treatment with a ns-NSAID, Sulindac, resulted in retardation of cysts development (Zhang et al., 2019). Higher COX-2 expression and no changes in COX-1 expression were noted in coronary arteries of renal hypertensive rats (Paula et al., 2018). COX-2 expression, but not COX-1, is markedly decreased in the cortex and increased in the inner medulla in response to unilateral ureteral obstruction (UUO) (Chou et al., 2003) and bilateral ureteral obstruction (BUO) (Cheng et al., 2004). Interestingly, the knockdown of COX-2 using chitosan/small interfering RNA (siRNA) nanoparticles prevented kidney injury induced by UUO in mice (Yang et al., 2015) (Table 2).
The cardio-renal effects of selective and nonselective NSAIDs under normal and pathological states are summarized below and in Table 3. The COX-1-inhibitory effects of NSAIDs on decreased platelet aggregation are well established (Campbell et al., 2007). However, the cardiovascular effects of s-NSAIDs in experimental laboratory studies have been variable due, in part, to variations in study design, genetic backgrounds, drug exposure, and the environment (Sellers et al., 2010). Under normal physiological conditions such as the case in toxicity studies which are conducted in normal laboratory animals, alterations in platelet aggregation would not likely be identified with COX-2 inhibition. Thus, pharmaceutical inhibition of COX-2 in toxicity studies in normal laboratory animals and animal models are not anticipated to cause significant reductions in PGI2 or TXA2 levels in vascular tissues. In human healthy volunteers, s-NSAIDs, such as celecoxib, had no effect on the anti-thrombotic effect of aspirin (Table 3) (Gladding et al., 2008; Wilner et al., 2002). In another human healthy volunteers study, celecoxib partially limited the angiotensin II-mediated increases in markers of oxidative stress (e.g., nitrotyrosine and nitric oxide metabolites) suggesting a favorable physiological pathway for improved cardiovascular risk profile (Pialoux et al., 2017). Pretreatment of naïve endothelial cells obtained from healthy volunteers with celecoxib resulted in protection from intermittent hypoxia-mediated injury (Khalyfa et al., 2017).
| Renal | s-NSAIDs | ns-NSAIDs |
|---|---|---|
| Normal physiologic state | No effects on GFR, electrolytes abnormalities and fluid retention | Increased GFR, electrolytes abnormalities and fluid retention, analgesic nephropathy, acute interstitial nephritis |
| Pathological state | Transient decreases in RBF and GFR in salt-depleted subjects, Lower renal adverse events and hospitalization in hypertension | Reduced GFR and renal plasma flow in polyarthritis |
| Cardiac | ||
| Normal physiologic state | Alterations in platelet aggregation is unlikely | Decreased platelet aggregation |
| Pathological state | Reduce myocardial infarction size, reduce ischemia/reperfusion injury, reduce progression of atherosclerotic lesions | Reduce or increase myocardial infarction size, reduce ischemia/reperfusion injury, reduced atherogenesis |
In control kidneys from mice, COX-1 expression before and after NSAID treatment was investigated (Meskell and Ettarh, 2011). Significant COX-1 expression was noted in the renal cortex and medulla with significantly more expression evident within the cortex. Expression in the superficial cortex was more pronounced than in the rest of the cortex or the medulla. COX-1 expression was expressed in the glomeruli and parietal cells of the glomerular capsule, epithelial cells of the proximal and distal convoluted tubules, and the collecting ducts. COX-1 expression within the distal tubule was present in the juxtaglomerular region, adjacent to the glomerulus. Interstitial cells in the renal cortex did not show COX-1 expression. Within the medulla, pronounced COX-1 expression of COX-1 was noted within the endothelial cells of the renal vasculature but the epithelial cells of the medullary collecting ducts and the interstitial cells in the medulla showed poor reactivity for COX-1 (Meskell and Ettarh, 2011; Khan et al., 2013). In the ns-NSAID, indomethacin, treated kidneys, COX-1 expression was present in the cortical and medullary areas. COX-2 null mice show postnatal developmental abnormalities in their kidneys that then progressively deteriorate with increasing age (Morham et al., 1995). In normal rats, celecoxib administration for up to 33 days partially prevented the increase in blood pressure, and this hemodynamic effect was associated with a reduction in plasma Angiotensin II (Guzmán-Hernández et al., 2015). In healthy cats, robenacoxib administration decreased urinary PGE2 excretion, but did not affect GFR; however, GFR was significantly decreased after ketoprofen administration (Pelligand et al., 2015). Robenacoxib has been shown to be safe in healthy dogs and cats receiving antihypertensive drugs and loop diuretics that could cause kidney injury (Kongara and Chambers, 2018). Impairment of vasodilatation in acute kidney injury (AKI) has been noted with NSAIDs (Radi, 2018, 2019). In healthy dogs, no effects on GFR were noted after administration of robenacoxib and no increased risk of AKI was noted with the combination of benazepril and robenacoxib (Panteri et al., 2017). In a study in healthy elderly volunteers placed on a fixed sodium intake, rofecoxib at 50 mg once daily dose for 14 days did not affect GFR and caused a transient retention of sodium, reduced sodium excretion by 20% during the first 3 days of drug administration and then returned to normal (Catella-Lawson et al., 1999). The renal effects of celecoxib and naproxen were compared in healthy elderly subjects 65 year of age and older (Whelton, 1999). Small transient decreases in urinary sodium excretion were observed with both NSAIDs. GFR was significantly reduced in the naproxen-treated volunteers while no significant effects on GFR were noted after treatment with celecoxib (Whelton, 1999) (Table 3).
Animal models of myocardial infarction (MI) are classified into four main categories: MI, ischemia with reperfusion (I/R), I/R with ischemic preconditioning, and I/R with post-conditioning (Sellers et al., 2010). The majority of animal models evaluating the cardiac effects of COX inhibition have utilized either MI or I/R with or without ischemic preconditioning (Sellers et al., 2010). The duration of the ischemic event prior to reperfusion affects the degree of tissue injury (Ferdinandy et al., 2007). The findings from COX inhibition in acute MI studies can be divided into short-term and long-term findings, with most studies are of less than 7 days in duration (Sellers et al., 2010). A short-term treatment with a s-NSAID, DFU (5,5-dimethyl-3-(3-fluorophenyl)-4-(4-methylsulphonyl) phenyl-2(5H)-furanon), in rats for 3 days and 3 months after coronary artery ligation revealed that DFU had smaller infarct size than controls, with better left ventricular function than vehicle-treated animals (Saito et al., 2004, 2003). Rats treated with the s-NSAID parecoxib for 5 days beginning 2 days post coronary artery occlusion had increased wall thickness at the site of infarction and improved cardiac function (Straino et al., 2007). Pigs treated with parecoxib for 7 days after MI have decreased apoptosis around the infarcted region. Pigs treated with aspirin (ASA) for 7 days before MI had no difference in infarct size or heart function. Those treated with a derivative of ASA, NCX 4016, however, did have reduced infarct size (Wainwright et al., 2002). Pigs treated for 6 weeks, after induction of MI, with celecoxib (plus clopidrogel and sotalol) had increased risk of mortality from left ventricular rupture at the infarction site. The area of infarction had decreased left ventricular wall thickness and decreased collagen within the infarcts, as well as decreased cardiovascular function (Timmers et al., 2007). Dogs treated with ASA, rofecoxib, or meclofenamate in acute MI studies had reductions in infarct size (Carnieto et al., 2009; Such et al., 1983) but long-term studies with ASA (6 weeks post-MI) showed no difference in infarct size from control animals (Brown et al., 1983). Dogs treated with ibuprofen for up to 24 hr or ASA for up to 3 hr after MI had increased scar thinning 6 weeks after MI (Brown et al., 1983). Primary rat cardiomyocytes subjected to hypoxia/reoxygenation (H/R) for 45 min have higher COX-2 expression and pretreatment with NS398, a s-NSAID, significantly attenuated H/R-induced cell injury (Pang et al., 2016). In rabbits, pretreatment with celecoxib, but not aspirin, for 3 days, significantly ameliorated the effects of acute MI (Zhao et al., 2012). Collectively, these studies suggest that COX-1 inhibition may have little impact on short and long-term effects of MI. COX-2 inhibition may reduce infarct size and inflammation at the site of MI acutely. The reduction in local inflammation may be acutely valuable but may have long-term adverse consequences in tissue breakdown and scar formation. Both selective and ns-NSAIDs may affect wound healing and clearing of bacterial infections following cardiovascular surgeries. While the findings after long-term inhibition of COX-2 after infarction are variable there is a trend in these studies indicating decreased collagen density and normal wall remodeling at the infarct site. This could result in scar thinning, which may adversely affect heart function (Sellers et al., 2010).
In multiple laboratory animal models (dogs, rats, and mice) which have undergone several cycles of ischemic preconditioning exhibited reduced myocardial injury when preconditioning was followed by prolonged ischemia with reperfusion (Alcindor et al., 2004; Ferdinandy et al., 2007; Guo et al., 2000; Sellers et al., 2010). The effect of ischemic preconditioning is lost if s-NSAIDs are administered prior to surgery, while the protective effect of preconditioning on the myocardium is not lost with pre-treatment with ASA (Alcindor et al., 2004; Guo et al., 2000). These studies demonstrated the importance of COX-2, not COX-1, in ischemic preconditioning associated myocardial protection. Although limited, many of the studies on the effect of COX inhibition in ischemia/reperfusion without preconditioning focus on the early/immediate effects of COX inhibition in reperfusion, rather than the long-term consequences in remodeling (Sellers et al., 2010). A study in dogs treated with rofecoxib prior cardiac ischemia followed by reperfusion showed no detectable differences in cardiac Troponin I (Carnieto et al., 2009), suggesting that acute injury is not altered in I/R injury with COX inhibition. A longer-term study in rats exposed to 8 hr of cardiac ischemia followed by 2 weeks of reperfusion and ASA therapy revealed no differences in infarct size, septal, or left ventricular wall thickness compared to vehicle-treated animals (Alhaddad et al., 1995). Collectively, I/R studies demonstrate that COX-2 is an important mediator in the protective effect of preconditioning in I/R studies. However, neither COX-1 nor COX-2 inhibition appears to significantly impact the outcome in I/R studies in several animal models (Sellers et al., 2010).
COX-1 inhibition appears to consistently modify the initiation and/or progression of atherosclerosis based on atherosclerotic mouse models (Belton et al., 2003; Cyrus et al., 2007, 2006; Sellers et al., 2010). In cholesterol-fed rabbits, a ns-NSAID, indomethacin, reduced the progression of atherosclerotic lesions and improved endothelium-mediated vascular responses (Srisawat et al., 2003). In a hypercholesterolemic pig model of chronic ischemia, both naproxen and celecoxib improved vessel relaxation and decreased the vasoconstrictive response to serotonin in the ischemic heart area (Chu et al., 2012). COX-2 inhibition with rofecoxib promoted early atherosclerotic lesion formation in low-density lipoprotein receptor (Ldlr)-deficient mice and COX-2 inhibition with Merck Frosst (MF) tricyclic increased both early atherosclerotic lesion area and atherosclerotic plaque destabilization in Apoe-deficient mice (Burleigh et al., 2005a; Rott et al., 2003). In Apoe-deficient mice, COX-2 inhibition, using SC-236, had no effect on lesion development or platelet interactions with vessel wall was noted (Belton et al., 2003). Chronic administration of celecoxib and MF-Tricyclic in mice did not influence or have an effect on the composition of advanced atherosclerotic lesions (Bea et al., 2003; Olesen et al., 2002; Praticò et al., 2001). Thus, data from animal models suggest that while COX-2 is highly expressed in atherosclerotic plaques, it likely plays a lesser role in lesion development and progression than does COX-1 (Sellers et al., 2010). The effects of COX inhibition on local blood flow in the coronary artery have been evaluated in dogs. In these studies, AA-induced coronary blood flow is attenuated by pretreatment with ASA, nimesulide, naproxen and celecoxib (Hennan et al., 2001; Hong et al., 2008). In contrast, acetylcholine (ACH)-induced coronary vasodilatation (nitric oxide-dependent pathway) in dogs was unaffected by pretreatment with naproxen and SC-58236, an experimental s-NSAID, did not significantly attenuate either AA or ACH-induced vasodilatation (Gross and Moore, 2004). These data suggest that the effects of AA, at least in the dog, are primarily COX-1 dependent (Sellers et al., 2010).
Blood pressure effects in preclinical species related to COX inhibition have been variable. Studies in normal animals indicate that ns-NSAIDs and/or s-NSAIDs have little effect on the systemic arterial blood pressure (Birck et al., 2000; Black et al., 1998; Brands et al., 2001; Manohar et al., 1996; Sellers et al., 2010). In normo-tensive human patients, blood pressure and renal function were not significantly altered by short-term treatment with standard doses (200 mg twice a day) of celecoxib (Dilger et al., 2002). While blood pressure studies in dogs appear to be consistent, there is reported variation between rat strains in the effects of COX-2 inhibition on blood pressure. A study compared the effects of diclofenac, celecoxib, and rofecoxib in Dahl salt sensitive rats found that after 8 weeks of treatment, celecoxib slightly (~ 4%), but significantly reduced systemic blood pressure as compared to controls and diclofenac and rofecoxib minimally but significantly increased systemic blood pressure (~1%) (Hermann et al., 2005). In addition, COX-2 inhibition in rats had no effect on blood pressure (Vanecková et al., 2005). In contrast, COX-2 inhibitors have been found to increase blood pressure in normo-tensive and hypertensive rats (Höcherl et al., 2002; Muscará et al., 2000). In a model of renovascular hypertension using Wistar-Kyoto or Sprague Dawley rats, COX-2 inhibition had no significant effect on blood pressure (Hartner et al., 2003; Richter et al., 2004). In a study of human patients taking anti-hypertensives, COX-2 inhibitors increased both systolic and diastolic blood pressure, but the effects on blood pressure were greater in people treated with rofecoxib than celecoxib (Whelton et al., 2001, 2002). Patients on angiotensin-converting enzyme (ACE) inhibitors treated with high doses of celecoxib had no statistically significant effects on blood pressure (White et al., 2002). Thus, COX inhibition may attenuate the effects of some anti-hypertensive therapeutics. The cardiovascular risk for events after using celecoxib or etoricoxib was compared with naproxen and no significant differences for either myocardial infarction or stroke were noted (De Vecchis et al., 2014). In another meta-analysis study, it was found that COX-2 selectivity may not play a role in the cardiovascular risk of NSAIDs (Gunter et al., 2017).
Nonselective NSAIDs can reduce glomerular filtration rate (GFR) by causing efferent arteriole constriction, and reducing renal blood flow (Dunn et al., 1988; Khan et al., 2013) and may aggravate renin-independent, sodium-sensitive hypertension, possibly in part by inhibition of the COX-1 responsible for sodium excretion (Okumura et al., 2002). NSAIDs contribute to changes in blood pressure and edema, resulting from renal flow and fluid retention imbalance. Inhibition of cortical and medullary PGs production by NSAIDs can lead to electrolytes abnormalities and fluid retention such as sodium, potassium and water retention (Khan et al., 2013). The effects of NSAIDs on urinary sodium and potassium excretion were studied in rats. Diclofenac and flurbiprofen were administered orally once daily for 4 days. Both diclofenac and flurbiprofen significantly reduced excretion rate of sodium and potassium (Harirforoosh and Jamali, 2005). The renal effect of ns-NSAID, sodium meclofenamate administered intravenously, in the pentobarbital-anesthetized and sodium-repleted dog was investigated. Sodium meclofenamate significantly reduced urinary excretion of sodium, but did not affect mean arterial blood pressure, renal blood flow (RBF), GFR, or urine volume (Blasingham and Nasjletti, 1980).
Analgesic nephropathy is characterized by chronic interstitial nephritis and renal papillary necrosis or calcifications. The hallmark of analgesic nephropathy is renal papillary necrosis (RPN) (Khan et al., 2013). Studies in laboratory animals (i.e., rats, dogs, rabbits) show these species are unusually susceptible to RPN, likely related to their unique renal anatomy and physiology compared to other species (Wiseman and Reinert, 1975; Khan et al., 1998; Radi, 2019). Thus, these laboratory animals are not necessarily a good model for predicting RPN in humans. Papillary interstitial cells are the first cell type to undergo morphological changes in NSAID-related RPN. These cells have limited proliferating capacity; therefore, repeated injury is irreversible and may result in a local loss of vasodilatory PGs and development of secondary ischemia (Khan et al., 1998). It has been speculated that the papillae of rats and dogs are susceptible to chemical injury because of the poor blood flow that predisposes to ischemia and accumulation of toxic chemicals (Khan et al., 2013).
Clinically detectable edema is easily managed in patients and readily reversible upon discontinuation of the use of NSAIDs. Four doses of naproxen (500 mg, administered twice daily) were administered every 12 hr in a double-blind crossover design. Naproxen significantly decreased the urinary excretion of water (19%), sodium (26%), and chloride (26%), and decreased osmolal clearance (18%). Plasma renin activity, aldosterone, or free water clearance did not change significantly with treatment (Eriksson et al., 1987). The effects of ibuprofen and indomethacin on renal function and electrolytes in the presence and absence of furosemide in healthy volunteers who are on a restricted sodium diet were evaluated (Passmore et al., 1990). Neither indomethacin (50 mg) nor ibuprofen (400 mg and 800 mg) affected renal blood flow, GFR or electrolyte excretion before furosemide. However, renal blood flow and GFR were significantly increased in the first 20 min after furosemide and these changes were significantly attenuated by indomethacin compared with placebo and ibuprofen 400 mg (Passmore et al., 1990). In a placebo-controlled, double-blind, cross-over design study, the effects of 7 day oral administration of naproxen at a 500 mg dose in the morning and 250 mg dose in the evening in patients with polyarthritis and stable impaired renal function were investigated (Eriksson et al., 1990). Naproxen reduced GFR and renal plasma flow by 18% and 13%, respectively, and plasma renin activity decreased by 38% during naproxen treatments. No significant change in plasma aldosterone was observed during treatment, but urinary aldosterone declined significantly by 34% and albuminuria decreased by 41%. No discernible effects on base excess, excretion of water, sodium, or potassium, or on osmolal clearance were noted. However, serum potassium increased slightly but significantly during naproxen treatment (Eriksson et al., 1990). The effect of a single dose of 500 mg of naproxen on renal function in healthy volunteers was studied. No effects on GFR were noted. Over the first 4 to 8 hr of the study, fractional excretion of sodium was reduced by approximately 50% (Dixey et al., 1987). Thus, naproxen should be used with caution in patients with fluid retention, hypertension, or heart failure.
In prelinical toxicity studies with celecoxib, transient sodium retention was reported in rats up through six weeks of treatment (Khan et al., 2013). In one study in rats, administration of celecoxib for 3 weeks resulted in a 30 mm Hg increase in systolic blood pressure while same dose administered to hypertensive rats increased systolic blood pressure by 33 mm Hg (Muscará et al., 2000). In another study, however, celecoxib did not alter blood pressure in hypertensive rats and it has been suggested that the natriuretic response to increased blood pressure may be preserved during inhibition of COX-2 (Radi and Khan, 2006). In a rat adjuvant-induced arthritis model, diclofenac but not celecoxib significantly enhanced blood pressure (Verhoeven et al., 2017). In rats administered NS-398 or meloxicam, responses to increased renal interstitial hydrostatic pressure, induced by direct renal interstitial volume expansion and fractional excretion of sodium, were preserved (Gross et al., 1999). In another study, COX-2 inhibition in rats showed no effect on systemic blood pressure (Vanecková et al., 2005). Beneficial effects of COX-2 inhibition on the renal system have been reported. In Wistar and stroke-prone spontaneously hypertensive (SHRSP) rats, the post-ischemic increase in fractional sodium excretion was blunted after celecoxib treatment (Knight and Johns, 2005). It was found that vascular dysfunction in the renal and intrarenal arteries in SHRSP rat model is prostanoid and Y chromosome dependent and COX inhibition abolishes vascular dysfunction (Khan et al., 2018). A study in rats found that celecoxib might reduce proteinuria in nephritic syndrome without impairing renal function (Lee et al., 2009). Streptozotocin-diabetic rats treated with SC-58236 showed a marked reduction in albuminuria, a reduction in kidney weight-to-body weight ratio, and transforming growth factor beta 1 (TGF-β) excretion and a marked decrease in the urinary excretion of tumor necrosis factor alpha (TNF-α) (Quilley et al., 2011).
In salt-depleted subjects, celecoxib was administered for 7 days at 200 mg and 400 mg twice daily doses. Celecoxib caused sodium and potassium retention and had no effect on systemic blood pressure (Rossat et al., 1999). Short-term and transient decreases in RBF and GFR were found at the highest dose of 400 mg on day 1 (Rossat et al., 1999). In another study, no individual age category of normotensive subjects had blood pressure or renal function effects significantly altered by short-term treatment with standard doses (200 mg twice a day) of celecoxib (Dilger et al., 2002). In ankylosing spondylitis and other spondyloarthritis patients, no major risk differences for serious cardio-renal adverse events were noted among exposure to s-NSAIDs (etoricoxib, celecoxib) and ns-NSAIDs (Kristensen et al., 2015).
A selective COX-2 inhibitor, rofecoxib, was withdrawn from the market in 2004, due to evidence of adverse CV outcomes in a placebo-controlled trial. It was widely debated whether these CV adverse outcomes were because of selective inhibition of COX-2 while sparing prostaglandins derived from COX-1. Because of this widespread controversy on potential CV safety profile of selective COX-2 inhibitors, a large randomized, multicenter, double-blind, non-inferiority trial involving patients who were at increased CV risk and had rheumatoid arthritis or osteoarthritis (PRECISION) was conducted (Nissen et al., 2016). This trial included more than 24,000 patients with a mean treatment duration of > 20 months. Patients were randomized to s-NSAID (celecoxib) or one of two ns-NSAIDs (ibuprofen or naproxen). The goal was to assess the noninferiority of celecoxib regarding the primary composite outcome of CV death (including hemorrhagic death), nonfatal MI, or nonfatal stroke. Secondary outcome measures included coronary revascularization or hospitalization for unstable angina or transient ischemic attack. In the intention-to-treat analyses, a primary outcome event occurred in 2.3% patients in the celecoxib group, 2.5% patients in the naproxen group, and in 2.7% patients in the ibuprofen group. Thus, fewer major adverse CV events occurred in the celecoxib group than in the ibuprofen group, but the difference did not reach significance in the intention-to-treat population (p=0.06). Similarly, the rates of renal adverse events and hospitalization for hypertension were significantly lower in the celecoxib group than in the ibuprofen group (p=0.002), although they did not differ significantly between the celecoxib and naproxen (p=0.19). The risk of gastrointestinal events was significantly lower with celecoxib than with naproxen (p=0.01) or ibuprofen (p=0.002) consistent with scientific rationale for development of this class of pharmaceutical drugs. Similarly, in another study, the ns-NSAID ibuprofen showed a consistent increase, compared with baseline, in systolic blood pressure (p value < 0.001), and a higher incidence of new-onset hypertension versus the s-NSAID celecoxib (Ruschitzka et al., 2017)
Taken together, the data from animal models and clinical experience indicate the cardio-renal safety of selective and non-selective NSAIDs is comparable. COX-mediated PGs are involved in renal functions and their inhibition is associated with potential adverse renal effects. The role of COX-mediated PGs on CV system remain unclear as transgenic models suggest some physiologic role of COX-2 mediated PGs but clinical CV outcomes of selective and ns-NSAIDs in PRECISION trial do not support these preclinical observations.
COX-1 and COX-2 derived prostaglandins are essential in maintaining various cardio-renal functions. Alterations in renal functions have been noted in both animals and humans given selective or non-selective NSAIDs. However, post-marketing withdrawal of certain s-NSAIDs due to concerns of CV complications has raised the question as to why no adverse cardiovascular findings were identified in preclinical toxicity studies. Chronic toxicity studies in rodents and non-rodents of up to 2 years in duration indicated that standard preclinical toxicology studies in multiple species at broad dose ranges did not uncover the putative adverse CV safety events identified in long-term clinical studies with some selective COX-2 inhibitors. Animal models of cardio-renal diseases and genetically engineered mice have provided some indirect insights in understanding potential role of cyclooxygenase-mediated PGs in homeostasis and disease. Long-term controlled clinical trials of selective and ns-NSAIDs do not suggest a differential effect on the cardio-renal system. Taking together, preclinical and clinical experience suggests the cardio-renal safety profile of selective and ns-NSAIDs is comparable.
The authors would like to acknowledge helpful review and feedback on the manuscript by Dr. Ikuo Horii, Pfizer Consultant, Tokyo, Japan, and Dr. Jon Cook, Pfizer, Groton, USA.
Conflict of interestThe authors are Pfizer employees.