Abstract
The endoplasmic reticulum (ER) adjusts its size and architecture to adapt to change in the surrounding environment. Russell bodies (RBs) were originally described as dilated structures of the ER cisternae containing large amounts of mutant immunoglobulin. Similar structures are observed in a wide variety of mutant proteins accumulated in the ER. We previously prepared Chinese hamster ovary (CHO) cells in which the expression of mutant antithrombin (AT) (C95R) was controlled with a Tet-On system and showed that RBs can be conditionally formed. However the precise architecture and intracellular behavior of RBs have been as yet only poorly characterized. To characterize the properties of RB, we prepared the same system using a green fluorescent protein (GFP)-fused mutant and measured the dynamics and architecture of RBs. We observed the mobile nature of the molecule in the RB lumen and RBs were separated from the rest of the ER network by narrow tubes. Furthermore, we found that the RBs were not simply expanded ER membranes. The RB lumen is filled with misfolded proteins that are surrounded by ER membranes. In addition, RBs mostly maintain their structure during cell division, possess ribosomes on their membranes and synthesize AT(C95R)-GFP. Based on the characterization of the hydrodynamic radius of AT(C95R)-GFP and the effect of DP1, an ER-shaping protein, we propose that RBs are spontaneously formed as a result of the partitioning of the misfolded AT with the shaping protein.
The endoplasmic reticulum (ER) is the site of synthesis and maturation of secretory proteins. The newly synthesized proteins are sequestered into the lumen of the ER and undergo various types of modification for proper maturation. Most of them are then packed into the coat protein complex II (COPII)-coated vesicles from ER exit sites (ERES) and undergo further modification at the Golgi apparatus. When they fail to obtain the correct conformation, they are mostly arrested at the ER, and a significant population is degraded by proteasome in the cytoplasm. A failure of proteasomal digestion leads to the accumulation of aberrant proteins in the cytosol. This process, collectively called ER-associated degradation (ERAD), is one of the critical steps for maintenance of cellular homeostasis.1) Although the process of transporting cargo across the membrane in a reverse form of protein synthesis has not been fully understood, the involvement of certain sets of membrane proteins associated with the ubiquitin ligase Hrd1 is firmly established.2–4) Various types of neurodegenerative diseases are associated with impaired function of this degradation.5)
On the other hand, if cargo proteins with some misfolded signatures are not properly disposed from the ER, the accumulation itself also disturbs ER homeostasis,6) and large, morphologically distinct structures often appear adjacent to the ER.7–9) Similarly, overexpression of membrane proteins often leads to the formation of “karmellae”-like or crystalloid structures. 3-Hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase is one of the first proteins whose overexpression phenotype was described in detail.10) Russell bodies (RBs) are designated as an inclusion body composed of the dilated ER cisternae containing condensed abnormal secretory proteins such as mutant immunoglobulins in plasma cells.8,11,12) Similar structures have also been observed in the accumulation of the mutant α1-antitrypsin expressed in hepatocyte13) and transgenic mouse liver14) or yeast expressing misfolded secretory proteins.15) The formation of RB is thought to be caused by the expansion of ER “stratum” that contains accumulated mutant proteins, as well as the isolation of aggregated mutant proteins. However little is known regarding the specific reasons for underlying RB formation.
The ER is a highly dynamic and amorphous organelle that can adjust its size and structure in response to changes in the intra- and extracellular environment.16) Its most characteristic shape is a tubular, web-like structure that spreads, particularly at the periphery of the cells. A series of recent findings has revealed the key molecules of this characteristic structure. At least two classes of membrane proteins, DP1 (YOP1 or REEP5) and reticulons are thought to be responsible for the formation of the narrow, unbranched tubules with a high curvature.17–20) Dynamic three-way junctions in the ER network are induced by Lnp1 and atlastins, a family of guanosine 5′-triphosphatases (GTPases).21) Another important structural feature, a large luminal volume of ER, appears to be maintained by a sheet structure. The low curvature of the sheet is generated by another membrane protein, Climp63.22)
Previously, we discovered a novel missense mutation, antithrombin (AT) Morioka (C95R), which causes the loss of one of the three disulfide bonds.23) Later, we reported that secretion of the mutant AT decreased ca. 1/50 times compared to that of the wild type AT and that the protein was largely not disposed of properly, but rather, accumulated in novel structures around the ER. This resembles the originally designated RBs.24,25) By controlling the expression under the tetracycline repressor, we showed the conditional RB-forming system.25) In this study, we visualized the expression by using green fluorescent protein (GFP)-tagged AT in the system and studied the structure and behavior, and the molecular basis of RB formation.
MATERIALS AND METHODS
MaterialsExpression vectors (pEGFP-N1 or pTRE2hyg), a cell line (CHO-K1 Tet-On), hygromycin B, and doxycycline were obtained from Clontech (Mountain View, CA, U.S.A.). Mouse anti-KDEL antibody, mouse anti-rat GM130 antibody, mouse anti-human LAMP2 antibody and rabbit anti-human AT antibody were obtained from Calbiochem (San Diego, CA, U.S.A.), BD Biosciences (Franklin Lakes, NJ, U.S.A.), Molecular Probes (Eugene, OR, U.S.A.), and Athens Research and Technology (Athens, GA, U.S.A.), respectively. Monoclonal antibody against rat β-tubulin was obtained from Sigma-Aldrich (St. Louis, MO, U.S.A.). Rabbit anti-GFP antibody was obtained from Molecular Probes (Eugene). Antibody against human ER-Golgi intermediate compartment-53 (ERGIC-53) was raised in rabbits and anti-rat Sec31 antibody was kindly provided by Dr. Kiyotaka Hatsuzawa (Tottori University, Tottori, Japan). Alexa Fluor 488 goat anti-rabbit immunoglobulin G (IgG) and Alexa Fluor 546 goat anti-mouse IgG were obtained from Molecular Probes (Eugene). The Effectene transfection reagent was obtained from Qiagen (Valencia, CA, U.S.A.).
Construction of Expression VectorsThe expression vectors of pcDNA3.1(+)/AT and pcDNA3.1(+)/AT(C95R) are described elsewhere.24) First to prepare expression vector for AT-GFP or AT(C95R)-GFP, cDNA fragments of wild type AT and mutant AT(C95R) were amplified using pcDNA3.1(+)/AT and AT(C95R) as the templates. Two oligonucleotides (the restriction sites are underlined), 5′-ATT ATA TTA TCTCGAGCCG CCA TGT ATT CCA ATG TGA TAG G-3′ and 5′-ATT ATA TTA TGGATCCGCC TTA ACA CAA GGG TTG GC-3′ were used as the forward and reverse primers, respectively. The polymerase chain reaction (PCR)-generated fragments were digested with XhoI and BamHI, then ligated into pEGFP-N1 expression vector at the corresponding sites, and designated pEGFP-N1/AT-GFP and pEGFP-N1/AT(C95R)-GFP, respectively. Then, to prepare an expression vector with the Tet-On system, cDNA fragments of wild type AT-GFP and mutant AT(C95R)-GFP were amplified using pEGFP-N1/AT-GFP and pEGFP-N1/AT(C95R)-GFP as the templates. Two oligonucleotides, 5′-ATT ATA TTA TGCGGCCGCCAT GTA TTC CAA TGT G-3′ and 5′-ATT ATA TAT AGTCGACTTA CTT GTA CAG CTC GTC CAT GC-3′ were used as the forward and reverse primers, respectively. The PCR-generated fragments were digested with NotI and SalI, subcloned in frame into pTRE2hyg expression vector, and designated pTRE2hyg/AT-GFP and pTRE2hyg/AT(C95R)-GFP, respectively.
Similarly, cDNA human DP1 was reverse-transcribed by Superscript II (Life Technologies Japan, Tokyo) with the primer 5′-TTG GTC GTA AGA AAC TGT GCC CAC-3′, or 5′-AAT AAT TAT ACC ACA GTC CC-3′ using total RNA isolated from HeLa cells, essentially as described.26) The DP1 cDNA was PCR-amplified with the primers 5′-ACT AGG AACAGATCTGAG CGG CGA GAC GG-3′ and 5′-GTAAAGCTTTTC AGC CCC ATT AGA AGG GCT CG-3′ and then subcloned into the BglII/HindIII sites of a plasmid for pmCherry-C1 using the In-Fusion HD cloning kit (TaKaRa Bio Inc., Shiga, Japan). The identity of all the clones was confirmed by DNA sequencing. The details on the construction of a plasmid for ss-cfSGFP2 (cysteine-free SGFP2 with a signal sequence of α1-antitrypsin) were reported previously.26)
Cell Culture, Establishment of Stably Expressing Cell Lines and TransfectionChinese hamster ovary (CHO) cells were cultured with Ham’s F-12 medium containing 10% (v/v) fetal bovine serum at 37°C and 5% CO2.24) To prepare CHO cells in which the expression of wild type AT-GFP or mutant AT(C95R)-GFP is controlled with doxycycline, CHO cells expressing Tet repressor protein were transfected with 3.0 µg of pTRE2hyg/AT-GFP and pTRE2hyg/AT(C95R)-GFP, respectively, using the Effectene™ transfection reagent. In the case of the CHO-K1 Tet-On cell line, the cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% (v/v) fetal calf serum, 100 µg/mL G418 and 100 µg/mL of hygromycin. The surviving 5–10 colonies were isolated by the cylinder technique and subjected to immunoblotting and fluorescence analysis of AT-GFP and AT(C95R)-GFP. Transfection of pmCherry-DP1 or pss-cfSGEP2 was performed as described previously.26)
Imaging and Mobility MeasurementsImmunostaining was performed as previously described.27) CHO cells expressing AT-GFP or AT(C95R)-GFP on coverslips were fixed with 4% formaldehyde for 5 min at room temperature or fixed with methanol at −20°C for 5 min. The primary antibodies used in this study were combinations of mouse antibodies against KDEL, GM130, LAMP2 and ERGIC-53. Cy3-conjugated goat anti-mouse IgG and Alexa Fluor 488 or 546 goat anti-mouse IgG, were used to decorate the primary antibodies. In some experiments, AT(C95R)-GFP was stained with the rabbit anti-GFP antibody followed by Alexa Fluor 488 goat anti-rabbit IgG. The cells were mounted in 90% glycerol in 100 mM Tris–HCl (pH 8.0), and the samples were examined by TCS-SP5 confocal microscopy (Leica Microsystems GmbH, Wetzlar, Germany) or LSM-510meta microscopy (CarlZeiss Oberkochen, Germany). The images of cells expressing mCherry-DP1 or ss-cfSGFP2 were captured with A1si (Nikon, Tokyo). GFP fluorescence of live cells was performed as previously described.28) CHO expressing wild type AT-GFP or mutant AT(C95R)-GFP cells were cultured on a glass-bottom dish in DMEM containing 10% (v/v) fetal calf serum, 100 µg/mL G418, 100 µg/mL hygromycin and 1 µg/mL doxycycline at 37°C in 5% CO2 containing atmosphere. The cells were observed with LSM510meta microscopy using a C-Apochromatic 40x (NA 1.2) or a Plan-Apochromat 63x (NA 1.4) objective lens.
For fluorescence recovery after photobleaching (FRAP) analysis, a 2 µm circle in the cell was photobleached, and the recovery was measured. Assuming that the majority of the process was caused by diffusion, we determined the characteristic diffusion time τdif by fitting the obtained recovery curve to the Soumpasis equation,29)
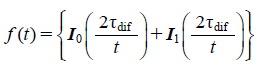 | (1) |
in which
I0 and
I1 are modified Bessel functions. The best-fit parameters were obtained by searching for numbers that minimalized the sum of the square of the residuals. For the calculation, we used the solver function of Excel2010 (Microsoft).
The hydrodynamic radius of AT(C95R)-GFP was determined by single-photon counting using PicoHarp300 (PicoQuant, Berlin, Garmany). The details were basically the same as reported26) except that the fluorescence light was split into two parts by a 50% beam splitter and detected by two single-photon avalanche diodes. The resulting two signals, F1 and F2, were cross-correlated to yield the normalized second order autocorrelation function, G(2)(τ), with the correlation time τ,
 | (2) |
in which
F1(
t) is the fluorescence intensity at time
t and the brackets denote time averaging. Assuming 3D Gaussian profile as the focal volume, the analytical model for the correlation function of free translational is
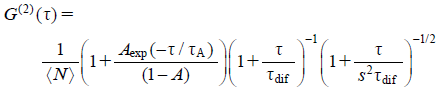 | (3) |
where ⟨
N⟩ is the average number of fluorophores in the confocal volume,
A the fraction of molecules in triplet state, τ
A the triplet time, τ
dif the diffusion time, and
s the structure parameter, which is the ratio of focal volume height to radius. When
n multi-components of diffusing properties are considered, the total correlation function is
Western BlottingWestern blot analysis was performed using primary antibodies and a secondary antibody, donkey anti-rabbit IgG antibody conjugated with horseradish peroxidase (Amersham Biosciences, Piscataway, NJ, U.S.A.). The antigen–antibody complex was visualized with ECL+ Western blotting detection reagent (Amersham Biosciences).
RESULTS
Time Course of the Appearance of RBsWe transfected pTRE2hyg/AT-GFP and pTRE2hyg/AT(C95R)-GFP into CHO K1 Tet-On cells, and established the CHO cell lines conditionally expressing wild type AT-GFP or the mutant AT(C95R)-GFP upon the addition of doxycycline (1 µg/mL) to visualize the dynamics of RB formation. The molecular mass of wild type AT-GFP and AT(C95R)-GFP was 85 kDa, close to the predicted size of GFP-tagged AT (Fig. 1A).
To monitor the formation of RB, we chased the cells in which AT(C95R)-GFP synthesis was triggered by the addition of doxycycline. The spherical structures accumulating AT(C95R)-GFP of ca. 0.5 µm appeared at approximately 24 h (Fig. 1B). We defined the structures as RBs in this study. The RB size increased at 48 h, ca. 70% CHO cells had more than one RB (Fig. 1C). Further incubation with doxycycline maintained the RBs (72 h). On the other hand, CHO cells expressing wild type AT-GFP did not form RBs but instead secreted into the medium (data not shown).
Subcellular Localization of AT(C95R)-GFP in CHO CellsTo clarify the specific localization of AT(C95R)-GFP in ER network as well as RBs in CHO cells, we examined the localization of AT(C95R)-GFP by immunofluorescence microscopy. As shown in Fig. 2A, the distribution of AT(C95R)-GFP was superimposable on the distribution of ER-resident proteins possessing the COOH-terminal KDEL sequence. The staining of RBs with the anti-KDEL antibody suggests that they were derived from the ER. Further, we examined the relationship of the RBs with other organelle markers by immunofluorescence microscopy. In contrast with the near complete co-localization with anti-KDEL antibody, RB rarely overlapped with other membrane markers such as the ER-Golgi intermediate compartment (ERGIC), lysosomes (LAMP2) or the Golgi apparatus (GM130) (Fig. 2B). These results suggest that almost AT(C95R)-GFP is retained in ER and RBs.
We next examined whether the ERES were included in the RBs. For this, using the anti-Sec31 antibody, we immunostained the cells incubated with doxycycline for 48 h. Sec31 is a component of the outer coat of COPII31) and has been used as a marker for ERES. As shown in Fig. 2C, some of RBs contained Sec31-positive spots. Apparently, ERES can be formed in RBs.
Precise Structure of RBsAs RBs seem to be large structures including ER membranes filled with AT(C95R)-GFP, we examined the precise structure of RB. First, RBs were sliced 0.4 µm intervals from bottom (b) to top (u) and continuous images were produced. Interestingly as shown in Fig. 3A, the bottom part (toward the dish) of the RB exhibited a rather reticular structure that condensed toward upper portion. AT(C95R)-GFP was co-localized to ER-resident proteins possessing the COOH-terminal KDEL sequence in the reticular structures in each section, and the structures became tight toward upper part of RB (Fig. 3B). In addition, Sec31 was detected inside of the RBs (Fig. 3C). The vertical confocal section revealed the presence of Sec31 from the bottom to the top in the RB (j). These results suggest that the RBs are not simply expanded ER membranes, but the RB lumen is filled with misfolded proteins that are surrounded by an ER membrane.
Dynamics of AT(C95R)-GFP in the ER Network and RBsTo analyze whether RBs are a part of the ER or are independent structures, we photobleached various regions of AT(C95R)-GFP positive structures including RBs and measured the recovery of GFP fluorescence in the region. By this method, the fluorescence recovery profile indicates information about the mobility as well as continuity of the structures containing the fluorescent protein. First, we measured the recovery of fluorescence in the CHO cells expressing AT(C95R)-GFP after 8 h incubation with doxycycline. As shown in Figs. 4A, B, when photobleaching was performed in the region, the fluorescence was rapidly recovered at a characteristic diffusion time of 2.15 s, indicating that AT(C95R)-GFP was mostly mobile at this stage (the maximum recovery rate was 0.77).
We also measured the mobility of AT(C95R)-GFP inside a single RB and between RBs. After 72 h incubation with doxycycline, several RBs were observed (Fig. 4C). When a small portion inside a given RB (indicated by circle) was photobleached, the fluorescence was recovered at a diffusion time of 2.52 s (Fig. 4E), which was only slightly slower compared to the recovery rate in the ER network after 8 h incubation with doxycycline (Fig. 4B). The maximum recovery rate of the fluorescence of these measurements was comparable (0.70), indicating the misfolded cargo remained mobile in RBs. However, when an entire region of a single RB (indicated by circle) was photobleached (Fig. 4D), little fluorescence was recovered (the maximum recovery rate was 0.028) even after 100 s. Only a marginal loss of fluorescence was observed in the RBs adjacent to the photobleached RBs.
To further confirm the confinement of the RB content apart from the rest of the ER, we performed fluorescence loss in photobleaching (FLIP) analysis. When an area of the ER network was photobleached every 1 min for 60 min, the fluorescence in the ER network close to the photobleached area was almost entirely lost, but the fluorescence in the RBs was not significantly changed even after 1 h (Fig. 4F). These results are consistent with FRAP, suggesting that RB content is hardly interchanged with the content of the surrounding structures.
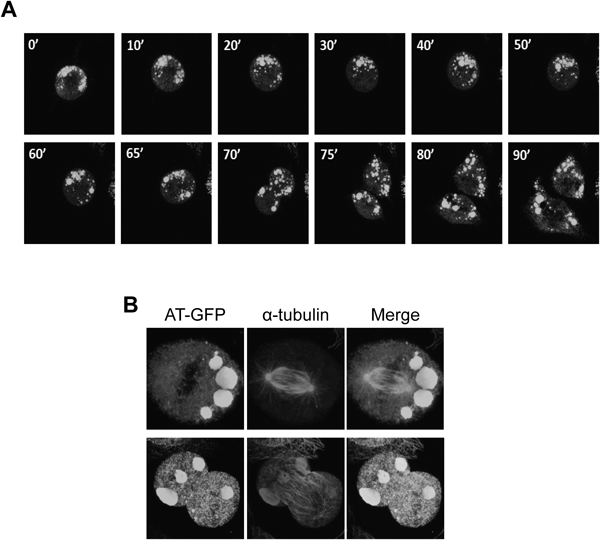
Dynamics of RB during Cell DivisionIt is well known that ER exhibits a dynamic network of interconnected sheets and tubules. In mitotic cells, the ER undergoes structural transformation of sheets into more fenestrated and tubular forms.16) To analyze whether RBs change the structural during cell division, we performed live-cell imaging of cells containing RBs. Figure 5A and the supplemental video show time-lapse images of cell division harboring RBs. During cytokinesis, no clear change of RB structures was observed. In this case, RBs were nearly equally divided into daughter cells. In Fig. 5B, there are images of immunostaining with anti-tubulin antibody. In the upper cells, spindle formation appeared to occur properly, whereas all the RBs were gathered into one daughter cell. In the lower case, on the other hand, one RB was separated. These results indicate that the RBs were randomly distributed to daughter cells without any structural change.
Biosynthesis of AT(C95R)-GFP on RBsAlthough RBs appear to form the isolated environment, ribosomes are attached to the RB membranes as in the rest of the ER.11,24) To investigate whether those ribosomes are functional and whether protein synthesis indeed occurs on the RBs of AT(C95R)-GFP, we photobleached the whole area of a cell containing RBs and then recorded the fluorescence. As shown in Fig. 6A, fluorescence of RBs was clearly recovered at 30 min post-bleaching. Quantitation of the fixed points in the RBs (spots 1 and 3) showed that newly synthesized AT(C95R)-GFP was continuously supplied into RBs. In the region where no RB was observed (spot 2), only a slight increase of intensity was observed. As the continuity between RB and the rest of the ER is very poor (Fig. 3), synthesis of AT(C95R)-GFP must be occurring in the RBs.
Involvement of ER Structure Forming Proteins in RB FormationRBs have generally been considered to be a stable large sheet with low membrane curvature, but Fig. 3 indicates that the entire RB structure may be constituted of tightly packed ER tubules or sheets. We speculate that AT(C95R) aggregates inside ER networks and the aggregates remain there and are segregated from other parts of the ER networks, then the ER membranes accumulating the aggregates are packed into RBs. Recent extensive studies have uncovered that a small number of membrane proteins determine the complicated ER structure.32) Among them, DP1 is able to self-assemble with lipids to form narrower tubules. DP1 is thought to be responsible for the formation of the narrow, unbranched tubules of high curvature.17–20) We examined whether the formation of narrow ER tubules by DP1 affects distribution of AT(C95R)-GFP in the cells. AT(C95R)-GFP synthesis was triggered by doxycycline. After 8 h, the cells were transfected with pmCherry-DP1, and the cells were observed under confocal microscopy 16 h after transfection. The result clearly showed that the majority of AT(C95R)-GFP was rather excluded from the periphery of cells and hardly detected in the extended reticular structure apparently induced by mCherry-DP1 overexpression (Fig. 7A). In a separate experiment, control CHO Tet-On cells were incubated with doxycycline for 8 h and then the cells were transfected with pmCherry-DP1 and pss-cfSGFP2, after which the cells were observed under confocal microscopy, as descried above. The image of fluorescent protein ss-cfSGFP2 (cysteine-free SGFP2 with a signal sequence of α1-antitrypsin) almost completely overlapped with that of mCherry-DP1 (Fig. 7B) and ss-cfSGFP2 was present in the tubules. The results suggest that aggregates of AT(C95R)-GFP are excluded from the narrow tubules of the ER network where DP1 was highly expressed.
The results shown in Fig. 7 are reminiscent of the exclusion of calreticulin from DP1-overexpressing cells.17) As this class of purified membrane proteins generated tubules of 15–20 nm in diameter, it was reasonable that AT(C95R)-GFP was excluded if the molecular mass was larger than the ER tubules generated by DP1. To test this, we prepared cell lysates and measured the hydrodynamic radius of AT(C95R)-GFP. In this experiment, we determined the cross-correlation of half-split fluorescence signal, which was obtained in two detectors detecting photons through a 50/50 beam splitter. The second-order correlation theoretically cancels triplet-derived decay in the correlation time and thus allows accurate estimate of diffusion time. The measurement showed that the correlation fitted well to the two components 3D diffusion model, as described in Materials and Methods (Figs. 8A–C) and the result showed that AT(C95R)-GFP contained a population with a 28.7 nm hydrodynamic radius of a large oligomer other than the 3.3 nm population (Fig. 8C), explaining the exclusion of the misfolded AT(C95R)-GFP from the DP1-enriched ER network.
DISCUSSION
The ER is composed of flattened sheets and tubules that branch to generate a polygonal network and plays a crucial role in the synthesis, modulation, transport and degradation of membrane and secretory proteins.1) The architecture of ER is also maintained through rapid fission and fusion of the ER network.33,34) RBs are thought to be membrane-bound structures derived from the ER, where misfolded proteins are accumulating.7–9) However, dynamics of RB and association of RB with other organelles are poorly understood. In the current study, we expressed GFP-tagged AT(C95R) under the control of the Tet-on system in CHO cells and clarified the intracellular architecture and dynamics of RBs in living cells.
Prior to the characterization of RB dynamics in the cells, we examined that subcellular localization of mutant AT(C95R)-GFP in CHO cells. The following evidence suggests that almost mutant AT(C95R)-GFP is located in ER network as well as RBs. The subcellular distribution pattern of AT(C95R)-GFP in CHO cells was superimposable on that of the ER resident proteins recognized by the anti-KDEL antibody (Fig. 2A). In contrast, the distribution of AT(C95R)-GFP was not consistent with the proteins located in the ERGIC, lysosomes and Golgi apparatus (Figs. 2B, C). Some mutant AT(C95R)-GFP might have been transported to ERGIC and Golgi apparatus via the secretory pathway, or degraded by proteasomes as well as lysosomes through autophagy. However, the AT(C95R)-GFP fluorescence was only scantly observed in these organelles, suggesting that the fluorescence emitted by AT(C95R) reflects the dynamics of the ER network and RBs in this study. In Figs. 2A, B, the ER resident proteins seem to have accumulated at the periphery of the RBs, while GM130, a Golgi marker seems to have merged with them in part. They might be due to the high expression of AT(C95R)-GFP.
When AT(C95R)-GFP was expressed in CHO cells by Tet-On system within 8 h, the ER has consecutive connection (probably normal) reticulation structures and did not form RBs at all. After photobleaching the ER network, recovery of the fluorescence was rapid (Fig. 4B). The recovery rate is essentially the same compared to other studies.28,35) Concerning the dynamics of the RBs, they began to appear after 24 h in the cells, with the size and number tending to increase over time.
At first we thought that RBs existed independently from ER network because of the following evidence. 1) The recovery of AT(C95R)-GFP fluorescence did not occur at all after the photobleaching of the entire RB, although immediate recovery of AT(C95R)-GFP fluorescence was observed when the area inside of the RB was photobleached (Figs. 4C, D). 2) According to FLIP analysis, the fluorescence in the ER network blocked off from the photobleached area was lost, but the fluorescence in RBs was not (Fig. 4F). These results also suggested that AT(C95R)-GFP diffused freely in the RBs. However, the detailed RB structure (Fig. 3) suggests that a part of the RB is connected with the ER network at the bottom of the RB. In addition, the size of misfolded AT(C95R)-GFP suggests it is unable to exit from pan-like structure of the RB. Therefore, we suggest that RB is a structure connected with the ER network via tubules.
In terms of a critical point addressed by the present study, the precise structure of the RB is unique. The tubular or sheet structures containing AT(C95R)-GFP were packed together so as to form a large, pan-like structure (Fig. 3). The structure was less compact in the bottom part and condensed toward the upper portion of the RB. The existence of Sec31 inside of the RB suggests that the structural elements derived from ER membranes were packed in the RB. The complex structure of RB was maintained during mitosis where fragmentation and reconstitution of the ER network occurred, suggesting the interaction among the tightly packed membranes might be kept well. In addition, we found that protein synthesis occurred in the RB (Fig. 6). Expansion of RBs including the reticular structures, is dependent on this protein synthesis, since the synthesis seems faster than that on ER network. However, it is not clear at present how the complex structures of RB form and why RB has a gradient of density as seen in Figs. 3B, C. In previous studies we observed ER expansion, but did not see any kinds of structures in the RB by electron micrographs.24,25) The reason for this discrepancy is unknown at present. A possible reason might be the conditions under which samples for electron micrographs were prepared. In other cases, membranous structures were observed inside of RB by the overexpression of mutant aspartic proteinase-I by immune-electron micrographs.15)
RBs accumulating AT(C95R) associated with the aggregation property of AT(C95R) as compared with that of other mutants such as AT(C8R) or AT(C128R) that were monomeric and did not result in the formation of RB-like structures.25) The fluorescence recovery rate of AT(C95R)-GFP at 72 h was slightly lower compared to 8 h (Fig. 4E) supports the aggregation property of AT(C95R)-GFP. Thus, we hypothesize that RBs containing AT(C95R)-GFP were spontaneously created by the segregation of ER network. Recent studies revealed a set of ER proteins determines the ER structures. DP1, an ER membrane protein shapes the tubular ER in mammalian cells.36) DP1 is highly enriched in the tubular portion of the ER and is involved in tubular formation of ER. If AT(C95R) accumulates as large cargo in the ER network, DP1, which preferentially resides in membranes of high curvature form clusters to avoid the large cargo from ER networks. As shown in Fig. 7, AT(C95R)-GFP was almost absent in the mCherry-DP1 positive peripheral structures. This effect seems to be created by a simple size exclusion of the narrow tubules. The fluorescent protein itself, whose hydrodynamic radius is approximately 2 nm (data not shown), entered the tubules (Fig. 7B), whereas AT(C95R)-GFP containing a population of more than 10-fold bigger molecules (Fig. 8) could not be diffused into such regions. It is known that some molecules like Climp63 or polysomes cannot enter in the tubules. We propose a possibility that AT(C95R)-GFP disturbs the normal transition of the ER between the sheet-rich structures and tubules, leading to the formation of RBs and eventually sequestrating AT(C95R)-GFP.
In this study, intracellular dynamics of RBs were characterized for the first time using living cells. We found that RBs are unique, stable structures that exist in manner that is segregated from the ER network. In addition, RBs have ribosomes and protein synthesis occurs on their membranes. RB formation might protect the cells against ER stress since the induction of ER chaperones such as Grp78, Grp97 and protein disulfide isomerase (PDI) was limited to a maximum of approximately two-fold under the conditions where expression of AT(C95R)-GFP was induced (data not shown). However, we cannot explain at least two of the current results at present, the first being why the sheets containing AT(C95R)-GFP are packed to form a large, pan-like structure. This must be another mechanism from the aforementioned spontaneous segregation model. In addition, it is not clear why some RB has a gradient of the sheet density as seen in Fig. 3. Further research is needed to clear up these points.
Acknowledgments
This research was supported in part by the Ministry of Education, Culture, Sports, Science and Technology of Japan (15659017 (TI), 20590054 (TI), and 40182969 (IW). Pacific Edit reviewed the manuscript prior to submission.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials. Video 1 shows the entire recorded images of Fig. 5A.
REFERENCES
- 1) Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem., 80, 71–99 (2011).
- 2) Kaneko M, Ishiguro M, Niinuma Y, Uesugi M, Nomura Y. Human HRD1 protects against ER stress-induced apoptosis through ER-associated degradation. FEBS Lett., 532, 147–152 (2002).
- 3) Kikkert M, Doolman R, Dai M, Avner R, Hassink G, van Voorden S, Thanedar S, Roitelman J, Chau V, Wiertz E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem., 279, 3525–3534 (2004).
- 4) Claessen JH, Kundrat L, Ploegh HL. Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol., 22, 22–32 (2012).
- 5) Scheper W, Hoozemans JJ. Endoplasmic reticulum protein quality control in neurodegenerative disease: the good, the bad and the therapy. Curr. Med. Chem., 16, 615–626 (2009).
- 6) Hegde RS, Ploegh HL. Quality and quantity control at the endoplasmic reticulum. Curr. Opin. Cell Biol., 22, 437–446 (2010).
- 7) Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol., 10, 524–530 (2000).
- 8) Kopito RR, Sitia R. Aggresomes and Russell bodies. Symptoms of cellular indigestion? EMBO Rep., 1, 225–231 (2000).
- 9) Schweitzer PA, Taylor SE, Shultz LD. Synthesis of abnormal immunoglobulins by hybridomas from autoimmune “viable motheaten” mutant mice. J. Cell Biol., 114, 35–43 (1991).
- 10) Chin DJ, Luskey KL, Anderson RG, Faust JR, Goldstein JL, Brown MS. Appearance of crystalloid endoplasmic reticulum in compactin-resistant Chinese hamster cells with a 500-fold increase in 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Proc. Natl. Acad. Sci. U.S.A., 79, 1185–1189 (1982).
- 11) Valetti C, Grossi CE, Milstein C, Sitia R. Russell bodies: a general response of secretory cells to synthesis of a mutant immunoglobulin which can neither exit from, nor be degraded in, the endoplasmic reticulum. J. Cell Biol., 115, 983–994 (1991).
- 12) Alanen A, Pira U, Colman A, Franklin RM. Mott cells: a model to study immunoglobulin secretion. Eur. J. Immunol., 17, 1573–1577 (1987).
- 13) Lomas DA, Li-Evans D, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature, 357, 605–607 (1992).
- 14) Carlson JA, Rogers BB, Sifers RN, Finegold MJ, Clift SM, DeMayo FJ, Bullock DW, Woo SL. Accumulation of PiZ alpha 1-antitrypsin causes liver damage in transgenic mice. J. Clin. Invest., 83, 1183–1190 (1989).
- 15) Umebayashi K, Hirata A, Fukuda R, Horiuchi H, Ohta A, Takagi M. Accumulation of misfolded protein aggregates leads to the formation of russell body-like dilated endoplasmic reticulum in yeast. Yeast, 13, 1009–1020 (1997).
- 16) Puhka M, Joensuu M, Vihinen H, Belevich I, Jokitalo E. Progressive sheet-to-tubule transformation is a general mechanism for endoplasmic reticulum partitioning in dividing mammalian cells. Mol. Biol. Cell, 23, 2424–2432 (2012).
- 17) Hu J, Shibata Y, Voss C, Shemesh T, Li Z, Coughlin M, Kozlov MM, Rapoport TA, Prinz WA. Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science, 319, 1247–1250 (2008).
- 18) Shibata Y, Voeltz GK, Rapoport TA. Rough sheets and smooth tubules. Cell, 126, 435–439 (2006).
- 19) Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell, 124, 573–586 (2006).
- 20) Hu J, Prinz WA, Rapoport TA. Weaving the web of ER tubules. Cell, 147, 1226–1231 (2011).
- 21) Chen S, Novick P, Ferro-Novick S. ER network formation requires a balance of the dynamin-like GTPase Sey1p and the Lunapark family member Lnp1p. Nat. Cell Biol., 14, 707–716 (2012).
- 22) Shibata Y, Shemesh T, Prinz WA, Palazzo AF, Kozlov MM, Rapoport TA. Mechanisms determining the morphology of the peripheral ER. Cell, 143, 774–788 (2010).
- 23) Ozawa T, Takikawa Y, Niiya K, Fujiwara T, Suzuki K, Sato S, Sakuragawa N. Antithrombin Morioka (Cys 95-Arg): a novel missense mutation causing type I antithrombin deficiency. Thromb. Haemost., 77, 403 (1997).
- 24) Tanaka Y, Ueda K, Ozawa T, Sakuragawa N, Yokota S, Sato R, Okamura S, Morita M, Imanaka T. Intracellular accumulation of antithrombin Morioka (C95R), a novel mutation causing type I antithrombin deficiency. J. Biol. Chem., 277, 51058–51067 (2002).
- 25) Tanaka Y, Ueda K, Ozawa T, Kitajima I, Okamura S, Morita M, Yokota S, Imanaka T. Mutation study of antithrombin: the roles of disulfide bonds in intracellular accumulation and formation of russell body-like structures. J. Biochem., 137, 273–285 (2005).
- 26) Suzuki T, Arai S, Takeuchi M, Sakurai C, Ebana H, Higashi T, Hashimoto H, Hatsuzawa K, Wada I. Development of cysteine-free fluorescent proteins for the oxidative environment. PLoS ONE, 7, e37551 (2012).
- 27) Sakurai C, Hashimoto H, Nakanishi H, Arai S, Wada Y, Sun-Wada GH, Wada I, Hatsuzawa K. SNAP-23 regulates phagosome formation and maturation in macrophages. Mol. Biol. Cell, 23, 4849–4863 (2012).
- 28) Kamada A, Nagaya H, Tamura T, Kinjo M, Jin HY, Yamashita T, Jimbow K, Kanoh H, Wada I. Regulation of immature protein dynamics in the endoplasmic reticulum. J. Biol. Chem., 279, 21533–21542 (2004).
- 29) Soumpasis DM. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys. J., 41, 95–97 (1983).
- 30) Gendron PO, Avaltroni F, Wilkinson KJ. Diffusion coefficients of several rhodamine derivatives as determined by pulsed field gradient-nuclear magnetic resonance and fluorescence correlation spectroscopy. J. Fluoresc., 18, 1093–1101 (2008).
- 31) Brandizzi F, Barlowe C. Organization of the ER-Golgi interface for membrane traffic control. Nat. Rev. Mol. Cell Biol., 14, 382–392 (2013).
- 32) Friedman JR, Voeltz GK. The ER in 3D: a multifunctional dynamic membrane network. Trends Cell Biol., 21, 709–717 (2011).
- 33) Kano F, Kondo H, Yamamoto A, Tanaka AR, Hosokawa N, Nagata K, Murata M. The maintenance of the endoplasmic reticulum network is regulated by p47, a cofactor of p97, through phosphorylation by cdc2 kinase. Genes Cells, 10, 333–344 (2005).
- 34) Pendin D, McNew JA, Daga A. Balancing ER dynamics: shaping, bending, severing, and mending membranes. Curr. Opin. Cell Biol., 23, 435–442 (2011).
- 35) Nagaya H, Tamura T, Higa-Nishiyama A, Ohashi K, Takeuchi M, Hashimoto H, Hatsuzawa K, Kinjo M, Okada T, Wada I. Regulated motion of glycoproteins revealed by direct visualization of a single cargo in the endoplasmic reticulum. J. Cell Biol., 180, 129–143 (2008).
- 36) Shibata Y, Voss C, Rist JM, Hu J, Rapoport TA, Prinz WA, Voeltz GK. The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J. Biol. Chem., 283, 18892–18904 (2008).