Abstract
Sophora flavescens AITON(kurara, 苦参)has long been used to treat various diseases. Although several research findings revealed the biosynthetic pathways of its characteristic chemical components as represented by matrine, insufficient analysis of transcriptome data hampered in-depth analysis of the underlying putative genes responsible for the biosynthesis of pharmaceutical chemical components. In this study, more than 200 million fastq format reads were generated by Illumina’s next-generation sequencing approach using nine types of tissue from S. flavescens, followed by CLC de novo assembly, ultimately yielding 83325 contigs in total. By mapping the reads back to the contigs, reads per kilobase of the transcript per million mapped reads values were calculated to demonstrate gene expression levels, and overrepresented gene ontology terms were evaluated using Fisher’s exact test. In search of the putative genes relevant to essential metabolic pathways, all 1350 unique enzyme commission numbers were used to map pathways against the Kyoto Encyclopedia of Genes and Genomes. By analyzing expression patterns, we proposed some candidate genes involved in the biosynthesis of isoflavonoids and quinolizidine alkaloids. Adopting RNA-Seq analysis, we obtained substantially credible contigs for downstream work. The preferential expression of the gene for putative lysine/ornithine decarboxylase committed in the initial step of matrine biosynthesis in leaves and stems was confirmed in semi-quantitative polymerase chain reaction (PCR) analysis. The findings in this report may serve as a stepping-stone for further research into this promising medicinal plant.
Sophora flavescens AITON(kurara, 苦参)has been recorded and used for more than 1800 years.1) As a widely distributed and effective herbal medicine, it severed as a cure for asthma, sores, gastrointestinal hemorrhage, diarrhea, allergy, inflammation in eastern Asian countries.2,3) Main chemical components of S. flavescens include flavonoids (1.5%), alkaloids (3.3%), alkylxanthones, quinones, triterpene glycosides, fatty acids as well as essential oils and recently, several clinical studies reported that alkaloids of S. flavescens were efficacious in treating various types of solid tumors (including breast, lung, liver and gastrointestinal tract cancers), which drew close attention to this traditional herbal plant.1,4)
In 1889, the characteristic compound matrine was isolated from the dry roots of S. flavescens by Nagai5) and then in 1966, Okuda et al. confirmed the absolute structure of (+)-matrine.6) Due to its notable medicinal efficacy, attempts to synthesize and biosynthesize matrine were conducted.7–9) In 1995, Saito et al. proposed the biosynthetic pathway of the carbon framework of matrine.10) Although the first steps of quinolizidine alkaloids biosynthesis have been elucidated recently in S. flavescens,11) more effort needs to be done to puzzle out the practical biosynthetic pathway of matrine.
S. flavescens also contain series of flavonoids and isoflavonoids such as kuraridin, kurarinone, isokurarinine, daidzein, maackiain.12) Much effort has been done to elucidate the biosynthetic pathway of flavonoids and many of the related genes are clear now in other species,13) but the ones involved in the biosynthesis of flavonoids in S. flavescens have not yet been discovered. In spite of several research findings cracking relevant quinolizidine alkaloids biosynthetic pathway and membrane-bound prenyltransferase,11,14,15) to our best knowledge, analysis using next generation sequencing approach for S. flavescens was not found up to date.
More and more genomes of model organisms have been sequenced.16,17) Nevertheless, for those non-model plants, lack of reference genome information jeopardizes studies on the underlying genes which are involved in biological processes related to vital plant physiology and drug development, etc. With this regard, transcriptome sequencing plays an essential role in apprehending the genetic diversity of organisms.18) Furthermore, such approaches help to obtain overall insights into whole gene sets associated to the protein diversity.19) Armed with well-established approaches such as Short Oligonucleotide Analysis Package (SOAP de novo),20) Assembly by Short Sequences (AbySS),21) Trinity,22) transcriptome profiling accelerates its pace in processing tremendous amount of data generated from large-scale sequencing projects. Despite the intensive challenges including library construction, reducing errors in image analysis and removal of low-quality reads, massively parallel cDNA sequencing (RNA-Seq) offers a more precise measurement of levels of transcripts and their isoforms than other methods.23)
In this study, 203598590 fastq format reads from 9 tissues of S. flavescens were generated by Illumina’s next-generation sequencing approach. CLC Genomics Workbench (CLC Bio, Denmark) was subsequently applied to conduct de novo assembly. Based on the findings provided by Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway mapping, the candidate genes that may be involved in the biosynthesis of key chemical components were identified. By studying expression pattern of genes related to the biosynthesis of quinolizidine alkaloids, we propose some promising contigs for future research.
MATERIALS AND METHODS
Plant Materials and Total RNA ExtractionExcept for leaf sample collected in May 2012, all the rest fresh tissues and organs were obtained from healthy S. flavescens plants growing in Chiba, Japan in June 2013. Callus tissue whose origin and subculturing were described previously,24) and other eight parts of the plant were sampled, including leaf, flower, stem, young bud, mature bud, bud right before blossom (BBB), pedicel while bud stage (PBS), pedicel while blossom (PWB) (Supplementary 1). The materials were dipped into RNA stabilization solution (RNAlater, Life Technologies, U.S.A.) immediately after removal from the field. Then RNAlater solution was gently wiped with a Kimwipe and the remaining sample was frozen by liquid nitrogen and subsequently powdered by Multi Beads Shocker (Yasui Kikai, Japan). By using TRIzol Reagent (Invitrogen, U.S.A.), total RNA was extracted from powdered tissues of S. flavescens, respectively. The obtained RNA was then cleaned up by RNeasy Mini Kit (Qiagen, U.S.A.).
For semi-quantitative reverse transcription-polymerase chain reaction (RT-PCR) analysis, in November 2014, the tissues of root, stem and leaf of kurara were sampled in the same location, where the other tissues were collected for deep-transcriptome sequencing, and subjected to the extraction of total RNA as described above.
cDNA Library Preparation and SequencingTruSeq RNA Sample Prep Kit v2 (Illumina, U.S.A.) was applied according to the manufacturer’s recommendation. Once the mRNA in total RNA had been polyA-selected and fragmented, double-stranded cDNA was prepared for cDNA library construction. With the creation of blunt-end fragments and the indexed adaptor ligation, the samples were hybridized to flow cells. Cluster amplification was performed using the cBot Cluster Generation System (Illumina, U.S.A.) and then sequenced by Illumina’s next-generation sequencing instrument.
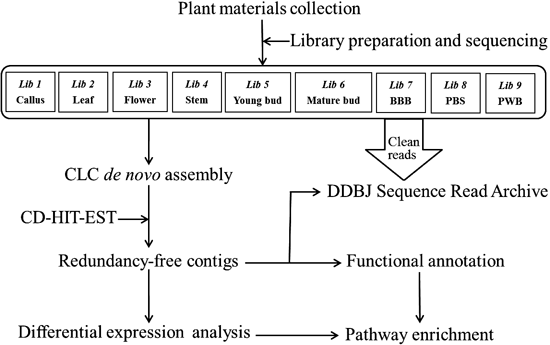
CLC Workbench for de Novo AssemblyPrior to assembly, the original fastq format data of S. flavescens were subjected to CLC trimming process in order to eliminate reads with poor quality and as a result, clean reads were obtained. The CLC approach (version 6.5) was then used to process the clean reads and all contigs over 300 bp were taken into consideration for downstream work. Since the assembly process may result in duplicate contigs due to sequencing error, CD-HIT-EST was utilized with representative sequences at 90% identity to obtain unique unigenes.25)
Reads per Kilobase of the Transcript per Million Mapped Reads (RPKM) Calculation and Expression-Based AnalysisThe standard formula for RPKM is as follows: RPKM=(109×C)/(N×L), where C is the number of reads mapped to the gene’s exons, N the total number of mapped reads in the experiment and L the total length of the exons in base pairs.26) In order to estimate the expression level of the contigs, with the aid of Burrows–Wheeler Aligner,27) Sequence Alignment/Map tools28) and High Throughput Sequencing (HTSeq),29) we applied the Mortazavi’s approach30) to map all the fastq format reads back to the contigs and calculated the RPKM values. Because the S. flavescens samples lacked technical replicates, the non-parametric approach for the identification of differentially expressed (DE) genes, NOISeq-sim,31) was adopted to analyze 36 independent pair-wise sample comparisons.
Annotation Pipeline and Data MiningAfter applying CD-HIT-EST, All 83325 de novo contigs were used as query sequences for the basic local alignment search tool (BLASTx) sequence similarity search against the non-redundant (NR) protein database at NCBI and the Universal Protein resource (UniProt) at UniProt consortium.32) The e-value threshold was set to 1e−10; the upper limit on the number of subject sequences from databases to show alignment was limited to 20. As to the large BLASTx output, only percent identities over 40% and e-values less than 1e−30 were taken into consideration. After eliminating redundancies, all unique gene identifiers in fasta format were uploaded to the UniProt ID mapping website for online data processing (http://www.uniprot.org). By consolidating the returned target list and the UniProtKB accession numbers (ACs) obtained from the above-mentioned BLASTx output against the UniProt database, we applied the redundancy-free ACs to annotation using the same online facilities. Out of the huge number of annotation results, we examined the reviewed findings from UniProtKB/Swiss-Prot as well as UniProtKB/TrEMBL for data mining. The sequences with Gene Ontology (GO) terms at the protein level were classified. Ultimately, 1350 enzyme commission (EC) numbers were applied to map pathways against the KEGG,33) and the enzymes related to flavonoids and isoflavonoids biosynthesis were studied.
Semi-quantitative RT-PCR for a Putative Lysine/Ornithine Decarboxylase (L/ODC) GeneBy using the total RNA extracted from the tissues harvested in 2014, cDNA was prepared according to the manufacturer’s instructions, SuperScript VILO kit (Invitrogen, U.S.A.). A 477 bp fragment of L/ODC was amplified by PCR using Ex taq DNA polymerase (TaKaRa, Japan) and specific primers (L/ODC-F: 5′-GAC ATT GGT GGC GGT TTC AC-3′, L/ODC-R: 5′-AGT GCT AAA GCC ATT GAA GTT GG-3′). The PCR for L/ODC cDNA was performed with an initial denaturation at 94°C for 2 min, then 26, 28 or 30 cycles each at 94°C for 30 s, at 54°C for 30 s, and 72°C for 50 s. For normalization of the different RNA preparations, a 571 bp fragment of S. flavescens β-actin was amplified with the following primers: (Act-F: 5′-AAG GCC AAC AGA GAG AAG ATG AC-3′, Act-R: 5′-ACC CAC CAC TAA GCA CGA TAT TT-3′). The PCR for β-actin cDNA was performed with an initial denaturation at 94°C for 2 min, then 22 cycles each at 94°C for 30 s, at 53°C for 30 s, and 72°C for 50 s. The PCR products were separated by electrophoresis using 1.5% gel at 100 V, and the gel was further stained by Ethidium Bromide Solution (Nacalai Tesque Inc., Japan).
RESULTS
Plant RNA Extraction and cDNA Library PreparationThe principal bioactive constituents of S. flavescens are the major quinolizidine alkaloids matrine and oxymatrine.34) The contents of these two components in S. flavescens are also the main valuation criteria of this plant. Meanwhile, considerable quantity of flavonoids and isoflavonoids are also accumulated in S. flavescens.35) In this study, we aimed to collect information about the nature of the genes responsible for the biosynthesis of matrine and oxymatrine, and study the related genes involved in the biosynthesis of flavonoids and isoflavonoids in S. flavescens. We extracted total RNA from the 9 tissues of this plant, resulting in 9 distinct cDNA libraries. We will refer to the libraries in the following manner: Lib 1 (callus), Lib 2 (leaf), Lib 3 (flower), Lib 4 (stem), Lib 5 (young bud), Lib 6 (mature bud), Lib 7 (bud right before blossom), Lib 8 (pedicel while bud stage), and Lib 9 (pedicel while blossom).
Illumina Sequencing and de Novo AssemblyAll 9 libraries were processed using the Illumina HiSeq platform. Empty reads, reads of low quality and those containing unknown bases were trimmed using CLC software. Nucleotide fastq sequences of Illumina reads were deposited at DDBJ Sequence Read Archive (DRA) with the accession number DRA003182. To practice de novo assembly, the fastq reads from all 9 libraries were combined together to generate fasta format contigs. After the entire run was assembled, the quality of the assembly (e.g., the ratio of aligned reads, average contig size, N75, N50 and N25 contig length) was identified to be better than the de novo results from individual libraries (data not shown). The assembly of the 9 libraries was utilized in the following analysis to discover their genetic information. 85054 contigs were generated. By applying CD-HIT-EST with a threshold of 0.9, duplicates were identified and removed, leaving 83325 non-redundant contigs. An overview of the experimental pipeline is shown in Fig. 1. Table 1 summarizes sequencing and assembly results.
Table 1. Overview of
S. flavescens Transcriptome Assembly
| Items | Numbers |
|---|
| Total bases | 20044281186 |
| Average length of reads (bp) | 98.4 |
| No. of reads | 203598590 |
| Average length of contigs (bp) | 664 |
| Maximum contig length (bp) | 15827 |
| N75, N50, N25 (bp) | 431, 969, 1947 |
| No. of contigs over 300 bp | 85054 |
| Non-redundant contigs | 83325 |
The reported GC content for unigene sequences in soybean and Arabidopsis was 43% and 44%, respectively.36) The average GC content of S. flavescens transcripts was found to be 39.3% (Supplementary 2). In eukaryotes, mean GC content varies from ca. 20 to 60%.37) Our values are in the middle of this range, slightly lower than those reported for Glycine max (43%) but very close to those reported for Medicago truncatula (40%).38)
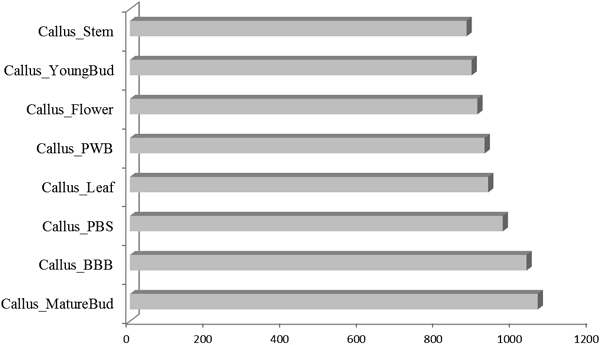
Transcriptome Information and the Differential Accumulation of TranscriptsRPKM calculation was performed as the first step of transcript expression analysis after RNA-Seq reads from every library were aligned to all contigs. RPKM calculation considers gene length variation and the number of total mapped reads, which allows this normalized output to be used directly for the comparison of gene expression. To perform DE analysis, 36 pair-wise comparisons of the nine libraries were conducted by applying NOISeq-sim, which is a non-parametric approach for the identification of DE genes from count data or previously normalized count data. By running NOISeq-sim on the R language platform with a given threshold (q=0.9) for selecting differentially expressed features, the resultant number of DE transcripts varied across comparisons. The highest value obtained was 1061 differences between callus and mature bud transcripts; the lowest value obtained was 0 between mature bud and BBB (Fig. 2 and Supplementary 3).
Protein Function Annotations and GO ClassificationFunctional annotations according to sequence similarity are often the initial step in studying the role and biological functions of gene products.39) BLAST program was utilized to scan nucleotide query sequences against protein databases (NR, UniProt) to identify similar subject sequences. When the threshold E-value for BLASTx searches was set to 1e−10 and if possible, the top 20 subject sequences for each query sequence were taken into consideration, we obtained 570611 subject sequences for all 83325 query sequences. To obtain reliable results while reducing redundancy, we set stricter requirements (refer to Annotation Pipeline and Data Mining) for retrieving the candidate genes. With this approach, significant matches were assigned to 27909 contigs.
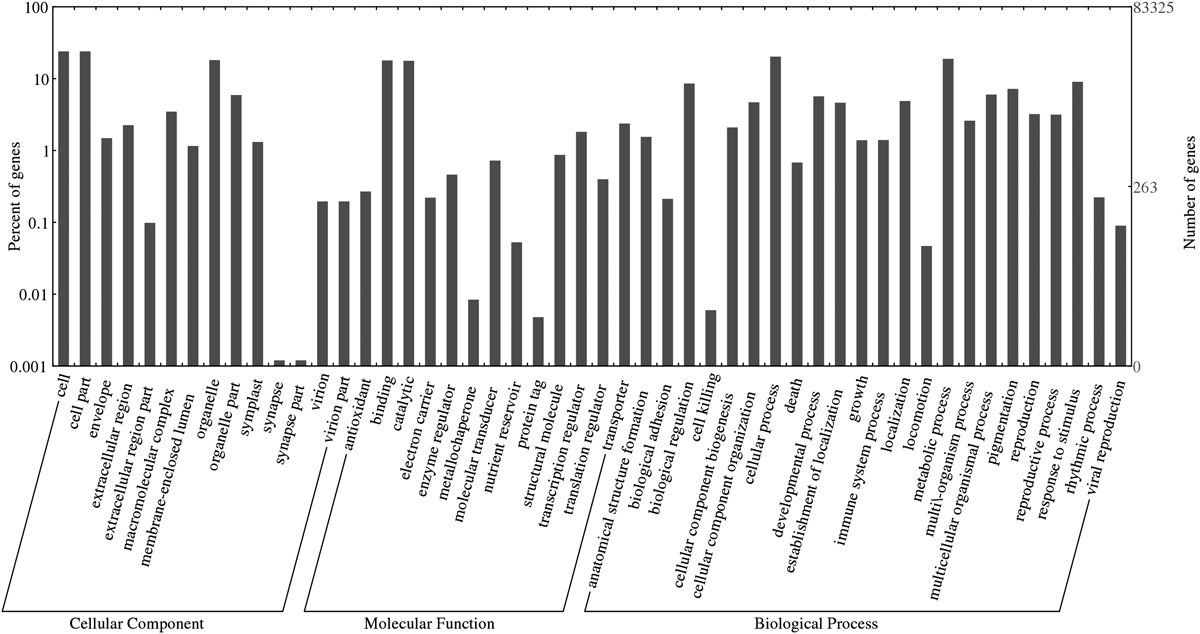
GO, consisting of three main domains (cellular components, molecular functions and biological processes), is a useful instrument with which to study the nature of annotated genes.40) Based on NCBI NR BLAST results, with the aid of Web Gene Ontology Annotation Plot (WEGO) software,41) 25921 contigs yielded corresponding GO terms that could be further classified into 50 sub-categories: 14 related to cellular components, 13 to molecular function and 23 to biological processes (Fig. 3). The limitation of evaluating gene classification by directly counting the number of GO terms which possess the same or very similar functions is that the expression level of these query sequences varies, which grants distinct weight to the same GO term as it corresponds to different query sequences. With this concern, overrepresented GO terms were identified by Fisher’s exact test. The one-tailed Fisher’s exact p-values corresponding to overrepresented categories were calculated according to the counts in 2×2 contingency tables. Counts n11, n12, n21, and n22 in each table stand for: n11, number of observations of a specific category in the first gene set; n12, number of other categories in the first gene set; n21, number of observations of a category in the second gene set; and n22, number of observations of other categories in the second gene set.42) p-Values were corrected using the false discovery rate (FDR) method with the threshold set at 0.05.43) For each S. flavescens library, contigs with RPKM value over 30.0 (the top ca. 10% of all transcripts) were regarded as highly expressed genes and extracted respectively. Then the merged 9867 contigs were used to perform Fisher’s exact test. The overrepresented GO terms (Supplementary 4, GO terms with p<1E−30 are listed) indicate expectedly cellular component like plasma membrane (GO: 0005886) and cytoplasm (GO: 0005737) plays crucial roles in all aspects regarding S. flavescens. Meanwhile, biological process including isopentenyl diphosphate biosynthetic process, methylerythritol 4-phosphate pathway (GO: 0019288) is also highlighted in the list, which will provide information for future research.
KEGG Pathway RetrievalKEGG44) provides a robust instrument for biological pathway assignment as well as the functional annotation of gene products. Based on ID mapping results, we obtained 1350 unique enzymes and subsequently retrieved pathways using KEGG. These EC numbers were assigned to 154 biological pathways with the largest number of enzymes (707) involved in metabolic pathways. Given the remarkable reputation of S. flavescens with regard to the ability to accumulate functional flavonoids, 18 flavonoid biosynthetic and 13 isoflavonoid biosynthetic enzymes are presented in Supplementary 5.
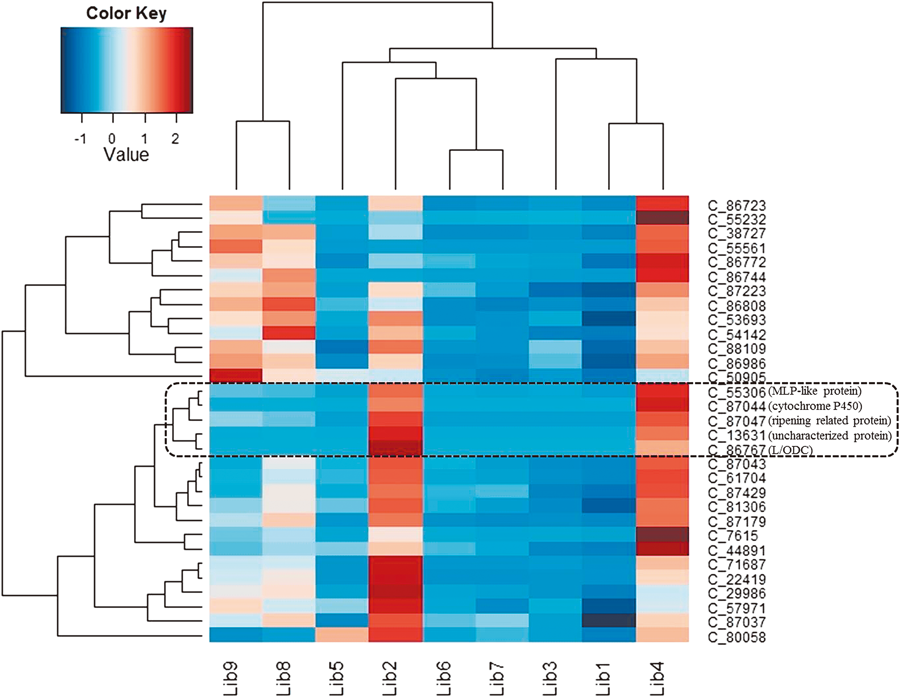
Putative Genes Involved in Isoflavonoid and Quinolizidine Alkaloids Biosynthesis in S. flavescensFlavonoids are a group of polyphenolic compounds distributed widely throughout the plant kingdom. These compounds modulate the activity of enzymes to benefit the entire organism. As an important subgroup of flavonoids, isoflavonoids are mainly produced in legumes and affect oxidative stress markers, immune function and adipogenesis.45) In the phenylpropanoid pathway, the synthesis of flavonoids is initialized by transforming phenylalanine into p-coumaroyl-CoA. To initiate flavonoid biosynthesis, chalcone synthase catalyzes the formation of chalcone scaffolds, from which all flavonoids derive.13,19) Based on our functional annotation findings, 34 contigs were predicted to represent seven enzymes critical to the biosynthesis of daidzein in S. flavescens. The number of contigs corresponding to each enzyme and the biosynthesis procedure are presented in Fig. 4. In our annotation result, contig c_86767 had 97.54% similarity to the L/ODC which catalyzes lysine into cadaverine and thus initiates the first steps toward the final product quinolizidine alkaloids including matrine and oxymatrine.11) By far, despite the sustained effort to crack the biosynthetic pathway regarding quinolizidine alkaloids, the identified genes underlying this process are very few. We focused on co-expression pattern related to c_86767 in order to hunt for candidate genes. According to RPKM values from all the studied organs, we calculate the Pearson correlation coefficients. The heatplot (Fig. 5) demonstrates some promising candidates including c_55306 (MLP-like protein), c_87044 (cytochrome P450) and c_87047 (ripening related protein). Such contigs shared very similar expression pattern across all the 9 tissues, suggesting their potential relationship in the process of quinolizidine alkaloid synthesis.
For contig c_86767 annotated as L/ODC gene, RPKM values of leaf and stem were as high as 553 and 321, respectively. Its expression pattern was further confirmed by semi-quantitative RT-PCR of L/ODC gene in the samples from leaf, stem and root of S. flavescence, using β-actin as the internal control gene,46) as shown in Supplementary 6. PCR amplification was carried out with three different cycle numbers (30, 28 and 26 cycles) to verify the linearity of semi-quantitative analysis. The highest expression was seen in leaf followed by stem. No detectable expression was observed in root in this condition. These RT-PCR results verified the expression pattern deduced from RPKM values provided by RNA-Seq analysis. Furthermore, it was also consistent with the preferential L/ODC expression in the leaf of Lupinus angustifolius.11) In all, accumulation of the final product such as matrine and oxymatrine would be high in other organs including root, but the initial steps for quinolizidine alkaloid biosynthesis feature in green parts of S. flavescens, such as leaf and stem as suggested previously.8,10)
DISCUSSION
S. flavescens has remarkable pharmaceutical attributes, including treating viral hepatitis, viral myocarditis, gastrointestinal hemorrhage and skin diseases (such as psoriasis and eczema). Numerous studies have reported the isolation and pharmacological action of the bioactive components in S. flavescens. Despite the importance and wide application of this medicinal plant, very few studies focused on its genetic profile. To generate overall deep transcriptome data for this plant will be helpful in discovering its underlying mechanism for the production of quinolizidine alkaloids. In this study, we utilized more than 200 million fastq format reads resulted from 9 tissues of S. flavescens to perform RNA-Seq analysis. 83325 representative contigs were obtained and the key enzymes involved in the biosynthetic pathways of active compounds were retrieved (Supplementary 5).
As to differentially expressed genes, NOISeq-sim output (Supplementary 3) showed DE genes identified by pair-wise comparison between callus and the rest 8 tissues were more than that of any other individual comparison. This is quite reasonable because in contrast with other organs of S. flavescens, callus consists of undifferentiated photoautotrophic cells. And in the slightly different situations as mature bud and bud right before blossom, not a single DE gene could be detected. Callus of S. flavescens does not accumulate quinolizidine alkaloids,8) which allows for the responsible genes to pop up by comparing it with other capable organs. By measuring the changes of matrine and oxymatrine in different growth stages47) and organs48) of S. flavescens by HPLC, the concentration of above-mentioned compounds in root and seeds was 3- to 6-fold higher than that of leaf, stem and flower. Nevertheless, such reports also showed leaf, stem and flower did accumulate considerable quantity of matrine and oxymatrine. These observations suggest that the gene(s) responsible for the formation of such characteristic compounds should lie in the list of differentially expressed genes (Fig. 2) and the alkaloids may translocate from the sites of de novo biosynthesis to the different sites of final accumulation.
Feeding studies illustrated the very first step for quinolizidine alkaloids biosynthesis. It concerns the decarboxylation of L-lysine into cadaverine by catalysis of L/ODC (EC 4.1.1.18). With the presence of copper amine oxidase (CuAO, EC 1.4.3.22), oxidative deamination of cadaverine produces 5-aminopentanal that spontaneously cyclizes to Δ1-piperideine Schiff base.49,50) The following steps to the final product matrine and oxymatrine still remain unknown. Based on co-expression pattern across the studied samples, we analyzed some candidate genes which were clustered into the same clade with the identified L/ODC gene. What’s more, the genes suggested by the RNA-Seq data of S. flavescens are also the ones indicated by the deep transcriptome data of Lupinus angustifolius, a species of legume that accumulates considerable quinolizidine alkaloids (Bunsupa et al., unpublished).
Using enzyme commission numbers identified in S. flavescens dataset to search KEGG for possible biosynthetic pathways retrieved 379 enzymes involved in biosynthesis of secondary metabolites. In addition to flavonoid biosynthetic pathway, valuable information about other pathways were also presented, such as monoterpenoid biosynthesis and steroid biosynthesis, which will contribute to the better understanding of the related mechanisms in plant kingdom. In this experiment, the obtained putative novel genes which may underlie the pharmaceutical function of S. flavescens and the findings on several essential biosynthetic pathways may serve as a stepping-stone for further studies on this promising and time-honored medicinal plant.
Acknowledgments
This study was supported, in part, by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan and by Health Labour Sciences Research Grant from the Ministry of Health, Labour and Welfare of Japan.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Sun M, Cao H, Sun L, Dong S, Bian Y, Han J, Zhang L, Ren S, Hu Y, Liu C, Xu L, Liu P. Antitumor activities of kushen: literature review. J. Evid. Based Complementary Altern. Med.: eCAM, 2012, 373219 (2012).
- 2) Hong MH, Lee JY, Jung H, Jin DH, Go HY, Kim JH, Jang BH, Shin YC, Ko SG. Sophora flavescens Aiton inhibits the production of pro-inflammatory cytokines through inhibition of the NF kappaB/IkappaB signal pathway in human mast cell line (HMC-1). Toxicol. In Vitro, 23, 251–258 (2009).
- 3) Funaya N, Haginaka J. Matrine- and oxymatrine-imprinted monodisperse polymers prepared by precipitation polymerization and their applications for the selective extraction of matrine-type alkaloids from Sophora flavescens AAITON. J. Chromatogr. A, 1248, 18–23 (2012).
- 4) Li LQ, Li XL, Wang L, Du WJ, Guo R, Liang HH, Liu X, Liang DS, Lu YJ, Shan HL, Jiang HC. Matrine inhibits breast cancer growth via miR-21/PTEN/Akt pathway in MCF-7 cells. Cell. Physiol. Biochem., 30, 631–641 (2012).
- 5) Nagai N. Study on Sophora flavescens. Yakugaku Zasshi, 84, 54–87 (1889).
- 6) Okuda S, Yoshimoto M, Tsuda K, Utzugi N. On the absolute configuration of matrin. Chem. Pharm. Bull., 14, 314–318 (1966).
- 7) Boiteau L, Boivin J, Liard A, Quiclet-Sire B, Zard Z. A short synthesis of (±)-matrine. Angew. Chem. Int. Ed., 37, 1128–1131 (1998).
- 8) Saito K, Yamazaki M, Yamakawa K, Fujisawa S, Takamatsu S, Kawaguchi A, Murakoshi I. Lupin alkaloids in tissue-culture of Sophora flavescens var. angustifolia—Greening induced production of matrine. Chem. Pharm. Bull., 37, 3001–3004 (1989).
- 9) Shibata S, Sankawa U. Biosynthesis of matrine. Chem. Ind., 1963, 1161–1162 (1963).
- 10) Saito K, Murakoshi I. Chemistry, biochemistry and chemotaxonomy of lupine alkaloids in the leguminosae. Studies in Natural Products Chemistry. (Atta ur R ed.) Vol. 15, Part C, Elsevier, pp. 519–549 (1995).
- 11) Bunsupa S, Katayama K, Ikeura E, Oikawa A, Toyooka K, Saito K, Yamazaki M. Lysine decarboxylase catalyzes the first step of quinolizidine alkaloid biosynthesis and coevolved with alkaloid production in leguminosae. Plant Cell, 24, 1202–1216 (2012).
- 12) Chen X, Yi C, Yang X, Wang X. Liquid chromatography of active principles in Sophora flavescens root. J. Chromatogr. B: Analyt. Technol. Biomed. Life Sci., 812, 149–163 (2004).
- 13) Falcone Ferreyra ML, Rius SP, Casati P. Flavonoids: biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci., 3, 222 (2012).
- 14) Gao T, Sun Z, Yao H, Song J, Zhu Y, Ma X, Chen S. Identification of Fabaceae plants using the DNA barcode matK. Planta Med., 77, 92–94 (2011).
- 15) Sasaki K, Tsurumaru Y, Yamamoto H, Yazaki K. Molecular characterization of a membrane-bound prenyltransferase specific for isoflavone from Sophora flavescens. J. Biol. Chem., 286, 24125–24134 (2011).
- 16) Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, Raney BJ, Pohl A, Malladi VS, Li CH, Lee BT, Learned K, Kirkup V, Hsu F, Heitner S, Harte RA, Haeussler M, Guruvadoo L, Goldman M, Giardine BM, Fujita PA, Dreszer TR, Diekhans M, Cline MS, Clawson H, Barber GP, Haussler D, Kent WJ. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res., 41 (D1), D64–D69 (2013).
- 17) Michael TP, Jackson S. The first 50 plant genomes. Plant Genome-Us, 6, 03.0001in (2013).
- 18) Muranaka T, Saito K. Phytochemical genomics on the way. Plant Cell Physiol., 54, 645–646 (2013).
- 19) Saito K. Phytochemical genomics—a new trend. Curr. Opin. Plant Biol., 16, 373–380 (2013).
- 20) Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics, 25, 1966–1967 (2009).
- 21) Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res., 19, 1117–1123 (2009).
- 22) Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol., 29, 644–652 (2011).
- 23) Fullwood MJ, Wei CL, Liu ET, Ruan Y. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res., 19, 521–532 (2009).
- 24) Yamamoto H, Kawai S, Mayumi J, Tanaka T, Iinuma M, Mizuno M. Prenylated flavanone production in callus-cultures of Sophora flavescens var. angustifolia. Z. Naturforsch. C, 46, 172–176 (1991).
- 25) Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28, 3150–3152 (2012).
- 26) Chen G, Li R, Shi L, Qi J, Hu P, Luo J, Liu M, Shi T. Revealing the missing expressed genes beyond the human reference genome by RNA-Seq. BMC Genomics, 12, 590 (2011).
- 27) Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 25, 1754–1760 (2009).
- 28) Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079 (2009).
- 29) Anders S. “Htseq: analysing high-throughput sequencing data with python.”: ‹http://www-huber.embl.de/users/anders/HTSeq/›, 2010.
- 30) Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods, 5, 621–628 (2008).
- 31) Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-Seq: A matter of depth. Genome Res., 21, 2213–2223 (2011).
- 32) Magrane M, Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database, 2011, bar009 (2011).
- 33) Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res., 40 (D1), D109–D114 (2012).
- 34) Ling JY, Zhang GY, Cui ZJ, Zhang CK. Supercritical fluid extraction of quinolizidine alkaloids from Sophora flavescens AAIT.. and purification by high-speed counter-current chromatography. J. Chromatogr. A, 1145, 123–127 (2007).
- 35) Shen Y, Feng ZM, Jiang JS, Yang YN, Zhang PC. Dibenzoyl and isoflavonoid glycosides from Sophora flavescens: inhibition of the cytotoxic effect of D-galactosamine on human hepatocyte HL-7702. J. Nat. Prod., 76, 2337–2345 (2013).
- 36) Tian AG, Wang J, Cui P, Han YJ, Xu H, Cong LJ, Huang XG, Wang XL, Jiao YZ, Wang BJ, Wang YJ, Zhang JS, Chen SY. Characterization of soybean genomic features by analysis of its expressed sequence tags. Theor. Appl. Genet., 108, 903–913 (2004).
- 37) Serres-Giardi L, Belkhir K, David J, Glemin S. Patterns and evolution of nucleotide landscapes in seed plants. Plant Cell, 24, 1379–1397 (2012).
- 38) Kawaguchi R, Bailey-Serres J. mRNA sequence features that contribute to translational regulation in Arabidopsis. Nucleic Acids Res., 33, 955–965 (2005).
- 39) Ramilowski JA, Sawai S, Seki H, Mochida K, Yoshida T, Sakurai T, Muranaka T, Saito K, Daub CO. Glycyrrhiza uralensis transcriptome landscape and study of phytochemicals. Plant Cell Physiol., 54, 697–710 (2013).
- 40) Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, Consortium GO, The Gene Ontology Consortium. Gene Ontology: tool for the unification of biology. Nat. Genet., 25, 25–29 (2000).
- 41) Ye J, Fang L, Zheng HK, Zhang Y, Chen J, Zhang ZJ, Wang J, Li ST, Li RQ, Bolund L, Wang J. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res., 34 (Web Server), W293–W297 (2006).
- 42) Takahashi H, Morioka R, Ito R, Oshima T, Altaf-Ul-Amin M, Ogasawara N, Kanaya S. Dynamics of time-lagged gene-to-metabolite networks of Escherichia coli elucidated by integrative omics approach. OMICS, 15, 15–23 (2011).
- 43) Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B: Met., 57, 289–300 (1995).
- 44) Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of genes and genomes. Nucleic Acids Res., 28, 27–30 (2000).
- 45) Miadoková E. Isoflavonoids—an overview of their biological activities and potential health benefits. Interdiscip. Toxicol., 2, 211–218 (2009).
- 46) Hong SM, Bahn SC, Lyu A, Jung HS, Ahn JH. Identification and testing of superior reference genes for a starting pool of transcript normalization in Arabidopsis. Plant Cell Physiol., 51, 1694–1706 (2010).
- 47) Zhang SR, Ji Y, Lin HM. Changes of oxmatrine and matrine in the development of Sophora flavescens AAIT.. Pratacultural Science, 25, 41–45 (2008).
- 48) Wang LL, Ma PQ, Pei CJ. Determination of oxymatrine content in different organs of Sophora flavescens AIT. by HPLC. Journal of Anhui Agri. Sci., 36, 5691–5692 (2008).
- 49) Bunsupa S, Yamazaki M, Saito K. Quinolizidine alkaloid biosynthesis: recent advances and future prospects. Front. Plant Sci., 3, 239 (2012).
- 50) Ma X, Gang DR. The lycopodium alkaloids. Nat. Prod. Rep., 21, 752–772 (2004).