Regular Articles
Synthesis of Clovamide Analogues That Inhibit NO Production in Activated BV-2 Microglial Cells
2017 Volume 40 Issue 9 Pages 1475-1482

Details
2017 Volume 40 Issue 9 Pages 1475-1482
A series of methyl ester of clovamide analogues, where the hydroxyl group of catechol moiety in caffeic acid and L-3,4-dihydroxyphenylalanine (L-dopa) was replaced with various functional groups, were synthesized and their inhibitory effects on nitric oxide (NO) production and inducible NO synthase (iNOS) expression in lipopolysaccharide (LPS)-induced BV2 cells were tested. Among the synthesized compounds, 3,5-ditrifluoromethyl analogue 9l (IC50=2.8 µM) exhibited a potency about 26.3 times greater than that of the parent compound 9a (IC50=73.6 µM) and suppressed NO production dose-dependently without cytotoxicity. Compound 9l also inhibited iNOS expression in LPS-induced BV2 cells at 2.5, 5 and 10 µM concentrations. These results suggested that the dihydroxyl group of catechol moiety in caffeic acid unit is not essential for the suppression of NO production and that 9l has potential as a potent inhibitor of NO production.
Nitric oxide (NO), a biological mediator in living organisms, plays an important role in cardiovascular, immune and neuronal systems. The highly reactive peroxynitrite anion (ONOO−), produced by the reaction of NO with superoxide anions, leads to oxidative damage in host tissue, modification of DNA bases, and disruption of enzyme function and structural proteins.1–3) Furthermore, the high level of NO production triggered by the transcriptional activation and production of inducible NO synthase (iNOS) in pathological situations has been considered an indicator of cytotoxicity in inflammation.4) Therefore, NO production is a critical step in numerous neurodegenerative diseases, and reduced NO production is a promising therapeutic strategy for the reduction of neuronal cell injury or death in various neuroinflammatory and neurodegenerative diseases.5–7)
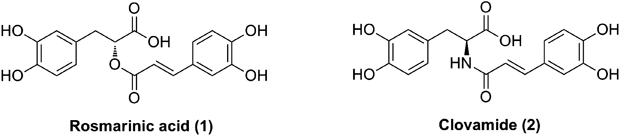
Clovamide, also known as N-caffeoyl-L-dihydroxyphenyl-alanine, is an amide isostere of rosmarinic acid and has two distinctive functional moieties (caffeic acid and L-dopa) linked by an amide bond8) (Fig. 1). Clovamide, which was originally identified in red clover (Trifolium pretense) and later discovered in cocoa liquor (Theobroma cacao), has been found in only a few plants in nature, but plays an important role in protecting plants against stress or pathogen attacks via oxidative enzyme as a part of defense system.9–12) In addition to the possession of anti-oxidant properties,13,14) clovamide also has a binding affinity for the p56lck SH2 domain, which is relevant to the treatment of immune disorders.15) Moreover, clovamide has anti-inflammatory effects via three different mechanisms (inhibition of superoxide anion production, pro-inflammatory cytokine release, and nuclear factor-kappaB (NF-κB) activation) in human monocyte cells,16) protective effects against oxidative stress in the rat cardiomyoblast cell line,17) as well as a neuroprotective effect in three different in vitro models of cell death (oxidative stress, excitotoxicity and oxygen-glucose deprivation (OGD)/reoxygenation).18) Although evidence suggested that clovamide can be an interesting plant-derived phenolic compound with expected beneficial effects, until now its physiological activity has not been adequately recognized because clovamide is not commercially available.
Most previous research related to clovamide was performed on clovamide or clovamide-type derivatives obtained directly by separation of extracts from plants. Only a few studies conducted with the synthesized analogues or derivatives, such as lipophilic clovamide-type derivatives such as N-caffeoyl tyrosine methyl ester, N-ferulyl L-dopa alkyl ester and N-sinapinyl L-dopa alkyl ester (alkyl=methyl, dodecyl), have revealed potent antioxidant activities in bulk lipids and moderated protectants in emulsions.8,19)
As part of our program to find a potent inhibitor of NO production, we recently found that clovamide derivative, 3-(3,4-dihydroxy-phenyl)-2-[4-(3-trifluoromethylphenyl)-but-2-enoylamino]-propionic acid methyl ester, revealed anti-inflammatory effect in lipopolysaccharide (LPS) stimulated rat derived primary cells, as well as murine BV-2 microglial cells.20) With the aim of improving the potency and elucidating the pharmacophore of clovamide, we modified clovamide by replacing the catechol group in caffeic acid and L-dopa with various functional groups. This paper provides details on the synthesis of various clovamide analogues and their inhibitory activities on NO production and iNOS expression on BV2 cells stimulated with LPS.
Melting points were determined on a Fisher-Johns melting point apparatus and were left uncorrected. 1H- and 13C-NMR spectra were recorded on a Varian Gemini 400 spectrometer at 400 and 100 MHz, respectively. The chemical shifts given are relative to tetramethylsilane. Infrared spectra were recorded on a Nicolet 6700 FT-IR spectrometer. Electrospray ionization (ESI)-MS spectra were obtained by Shimadzu LCMS-2010EV. Column chromatography was carried out using Merck silica gel 60 (230–400 mesh). All reactions were performed under a nitrogen atmosphere.
Methyl (2S)-2-Amino-3-(3,4-dihydroxyphenyl)propanoate Hydrochloride (5a)To a solution of L-dopa (10.0 g, 50.7 mmol) in MeOH (300 mL) was added thionyl chloride (11.1 mL, 152 mmol) dropwise. The mixture was heated to reflux temperature for 8 h. After the solution was cooled to room temperature, the solvent was evaporated in vacuo to obtain the title compound as a white foam (12.0 g, 95.2%). 1H-NMR (dimethyl sulfoxide (DMSO)-d6) δ: 2.88–2.95 (1H, m), 2.98–3.04 (1H, m), 3.64 (3H, s), 4.04–4.10 (1H, m), 6.45–6.50 (1H, d), 6.40–6.50 (1H, d, J=8.0 Hz), 6.59 (1H, s), 6.66–6.70 (1H, d, J=7.6 Hz); 13C-NMR (DMSO-d6) δ: 35.34, 52.55, 53.45, 115.64, 116.55, 119.98, 124.67, 144.42, 145.06, 169.18.
Methyl (2S)-2-Amino-3-(3,4-difluorophenyl)propanoate Hydrochloride (5b)To a solution of (S)-2-((tert-butoxycarbonyl)amino)-3-(3,4-difluorophenyl)propanoic acid (550 mg, 183 mmol) in 1,4-dioxane (10 mL) was added 4 N HCl in 1,4-dioxane (5 mL). After the mixture was stirred at room temperature for 4 h, hexane (10 mL) was added and stirred an additional 0.5 h. The resulting solid was filtered in vacuo and dissolved in MeOH (30 mL) was added thionyl chloride (397 µL, 152 mmol) dropwise. The mixture was heated to reflux temperature for 8 h. After the solution was cooled to room temperature, the solvent was evaporated in vacuo to obtain the title compound as a white foam (330 mg, 71.9%). 1H-NMR (CDCl3) δ: 2.70–2.80 (1H, m), 2.90–3.00 (1H, m), 3.55–3.63 (1H, m), 3.64 (3H, s), 6.80–6.88 (1H, m), 6.92–7.05 (2H, m); 13C-NMR (CDCl3) δ: 40.13, 52.09, 55.62, 116.96 (d, JC–F=16.7 Hz), 117.84 (d, JC–F=16.6 Hz), 125.04, 134.12, 147.88 (d, JC–F=244.9 Hz), 148.63 (d, JC–F=245.8 Hz); MS m/z: 216.00 ([M+H]+).
General Procedure for the Synthesis of Compounds 7a and b and 9a–lTo a solution of the corresponding substituted acid (1 eq, 2.02 mmol) in dichloromethane–N,N-dimethylformamide (DMF) (3 : 1, 20 mL) was added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (1 eq, 2.02 mmol), N,N-diisopropylethylamine (DIEA) (1 eq, 2.02 mmol) and N-hydroxybenzotriazole (HOBt) (1 eq, 2.02 mmol). After the mixture was stirred at room temperature for 30 min, corresponding amine (1 eq, 2.02 mmol) and triethylammonium acetate (TEA) (3 eq, 6.06 mmol) was added. The solution was stirred at room temperature for 4 h and then extracted with dichloromethane. The organic layer was washed with 1 N HCl solution, saturated NaHCO3 solution, water and brine, dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (ethyl acetate–hexane, 1 : 2) to obtain the title compound.
Methyl (2S)-2-((2E)-3-(3,4-Difluorophenyl)acrylamido)-3-(3,4-dihydroxyphenyl)propanoate (7a)White foam (400 mg, 52.5%); IR (KBr) cm−1: 3358, 1734, 1620; 1H-NMR (DMSO-d6) δ: 2.76–2.94 (2H, m), 3.62 (3H, s), 4.46–4.56 (1H, m), 6.42–6.48 (1H, d, J=8.4 Hz), 6.56–6.62 (2H, m), 6.62–6.70 (1H, d, J=14.8 Hz), 7.32–7.50 (3H, m), 7.60–7.68 (1H, t, J=10.4 Hz); 13C-NMR (DMSO-d6) δ: 36.43, 51.95, 54.22, 115.34, 116.14, 116.25, 117.850 (d, JC–F=17.4 Hz), 119.72, 122.67, 124.69, 127.61, 132.54, 137.27, 143.83, 144.85, 148.21 (d, JC–F=243.4 Hz), 148.55 (d, JC–F=247.2 Hz), 164.44, 171.94; MS m/z: 378.10 ([M+H]+).
Methyl 3-(3,4-Difluorophenyl)-(2S)-2-((2E)-3-(3,4-dihydroxyphenyl)acrylamido)propanoate (7b)White foam (50 mg, 55.6%); IR (KBr) cm−1: 3352, 1744, 1669; 1H-NMR (DMSO-d6) δ: 2.90–2.98 (1H, m), 3.04–3.12 (1H, m), 3.62 (3H, s), 4.56–4.68 (1H, m), 6.40–6.45 (1H, m), 6.70–6.78 (1H, m), 6.80–6.88 (1H, m), 6.92–7.00 (1H, m), 7.05–7.12 (1H, m), 7.20–7.28 (1H, m), 7.30–7.40 (2H, m), 8.40–8.58 (1H, m), 9.20 (1H, s), 9.39 (1H, s); 13C-NMR (DMSO-d6) δ: 35.83, 51.98, 53.40, 113.74, 115.63, 116.91 (d, J=16.7 Hz), 117.21, 117.88 (d, J=16.7 Hz), 120.52, 125.79, 125.94, 134.91, 140.03, 145.36, 146.86 (dd, J=69.7, 12.1 Hz), 147.37, 149.28 (dd, J=70.6, 12.1 Hz), 165.24, 171.69; MS m/z: 378.12 ([M+H]+).
Methyl 3-(3,4-Dihydroxyphenyl)-(2S)-2-((2E)-3-(3,4-dihydroxyphenyl)acrylamido)propanoate (9a)White foam (250 mg, 33.2%); IR (KBr) cm−1: 3397, 1728, 1603; 1H-NMR (DMSO-d6) δ: 2.70–2.80 (1H, m), 2.82–2.90 (1H, m), 3.59 (3H, s), 4.40–4.50 (1H, m), 6.34–6.41 (1H, d, J=15.6 Hz), 6.42–6.48 (1H, dd, J=8.0, 2.0 Hz), 6.53–6.62 (2H, m), 6.70–6.74 (1H, d, J=8.0 Hz), 6.75–6.83 (1H, dd, J=8.0, 1.6 Hz), 6.92 (1H, s), 7.10–7.25 (1H, d, J=15.2 Hz), 8.30–8.40 (1H, d, J=7.2 Hz); 13C-NMR (DMSO-d6) δ: 36.44, 51.79, 54.12, 113.71, 115.23, 115.59, 116.17, 117.50, 119.60, 120.41, 126.00, 127.66, 139.74, 143.73, 144.75, 145.31, 147.27, 165.12, 172.11; MS (ESI) m/z 374.10 ([M+H]+).
Methyl 3-(3,4-Dihydroxyphenyl)-(2S)-2-((2E)-3-(3-hydroxyphenyl)acrylamido)propanoate (9b)White foam (300 mg, 41.6%); IR (KBr) cm−1: 3358, 1723, 1598; 1H-NMR (DMSO-d6) δ: 2.72–2.80 (1H, m), 2.84–2.92 (1H, m), 3.60 (3H, s), 4.44–4.52 (1H, q, J=4.0 Hz), 6.40–6.48 (1H, d, J=8.0 Hz), 6.54–6.60 (2H, m), 6.60–6.64 (1H, m), 6.72–6.80 (1H, d, J=8.0 Hz), 6.89 (1H, s), 6.94–6.98 (1H, d, J=7.2 Hz), 7.16–7.22 (1H, t, J=7.2 Hz), 7.24–7.32 (1H, d, J=15.2 Hz); 13C-NMR (DMSO-d6) δ: 36.44, 51.89, 54.18, 113.65, 115.27, 116.19, 116.72, 118.65, 119.65, 121.02, 127.60, 129.82, 135.80, 139.49, 143.77, 144.79, 157.45, 164.72, 171.99; MS m/z: 358.15 ([M+H]+), 396.10 ([M+K]+).
Methyl 3-(3,4-Dihydroxyphenyl)-(2S)-2-((2E)-3-(4-hydroxyphenyl)acrylamido)propanoate (9c)White foam (250 mg, 34.7%); IR (KBr) cm−1: 3347, 1723, 1598; 1H-NMR (DMSO-d6) δ: 2.70–2.80 (1H, m), 2.84–2.90 (1H, m), 3.60 (3H, s), 4.44–4.50 (1H, m), 6.42–6.50 (2H, m), 6.56–6.62 (2H, m), 6.74–6.82 (2H, d, J=8.0 Hz), 7.24–7.32 (1H, d, J=16.0 Hz), 7.36–7.40 (2H, d, J=8.0 Hz); 13C-NMR (DMSO-d6) δ: 36.42, 51.79, 54.09, 115.22, 115.60, 116.15, 117.65, 119.60, 125.53, 127.63, 129.14, 139.34, 143.71, 144.74, 158.71, 165.71, 165.10, 172.07; MS m/z: 358.20 ([M+H]+), 396.15 ([M+K]+).
Methyl 3-(3,4-Dihydroxyphenyl)-(2S)-2-((2E)-3-(3-fluorophenyl)acrylamido)propanoate (9d)White foam (280 mg, 38.6%); IR (KBr) cm−1: 3342, 1734, 1608; 1H-NMR (DMSO-d6) δ: 2.74–2.84 (1H, m), 2.86–2.94 (1H, m), 3.61 (3H, s), 4.46–4.56 (1H, m), 6.42–6.70 (1H, m), 6.58–6.64 (2H, m), 6.70–6.78 (1H, d, J=15.6 Hz), 7.16–7.24 (1H, t, J=8.8 Hz), 7.36–7.48 (4H, m); 13C-NMR (DMSO-d6) δ: 36.41, 51.94, 54.19, 113.80 (d, JC–F=22.0 Hz), 115.29, 116.21, 119.66, 122.83, 123.64, 127.55, 130.73, 130.81, 137.12, 137.20, 143.81, 144.82, 161.00 (JC–F=241.9 Hz), 163.41, 164.42, 171.93; MS m/z: 360.20 ([M+H]+).
Methyl 3-(3,4-Dihydroxyphenyl)-(2S)-2-((2E)-3-(4-fluorophenyl)acrylamido)propanoate (9e)White foam (300 mg, 41.1%); IR (KBr) cm−1: 3352, 1745, 1669; 1H-NMR (CDCl3) δ: 2.93–3.05 (2H, m), 3.63 (3H, s), 4.80–4.90 (1H, m), 6.15–6.28 (1H, d, J=16.0 Hz), 6.35–6.50 (2H, m), 6.65–6.75 (1H, d, J=8.0 Hz), 6.90–6.98 (2H, t, J=8.8 Hz), 7.25–7.35 (2H, m), 7.40–7.50 (1H, d, J=15.2 Hz); 13C-NMR (CDCl3) δ: 36.83, 52.53, 53.73, 115.25, 115.55 (d, JC–F=22.0 Hz), 116.00, 119.07, 120.95, 127.45, 129.57, 130.40, 140.76, 143.39, 144.07, 162.07 (d, JC–F=249.5 Hz), 165.96, 172.15; MS m/z: 360.30 ([M+H]+).
Methyl 3-(3,4-Dihydroxyphenyl)-(2S)-2-((2E)-3-(3-methoxyphenyl)acrylamido)propanoate (9f)White foam (400 mg, 53.4%); IR (KBr) cm−1: 3348, 1734, 1641. 1H-NMR (DMSO-d6) δ: 2.75–2.81 (1H, m), 2.86–2.92 (1H, m), 3.62 (3H, s), 3.78 (3H, s), 4.45–4.55 (1H, m), 6.42–6.50 (1H, m), 6.58–6.64 (2H, m), 6.66–6.74 (1H, d, J=15.6 Hz), 6.92–6.98 (1H, m), 7.10–7.16 (2H, m), 7.30–7.40 (2H, m), 8.42–8.50 (1H, d, J=8.0 Hz); 13C-NMR (DMSO-d6) δ: 36.41, 51.86, 55.09, 112.38, 115.34, 116.16, 119.60, 119.85, 121.59, 127.55, 129.81, 135.95, 139.17, 143.75, 144.77, 159.30, 164.62, 171.93; MS m/z: 371.14 ([M+H]+).
Methyl 3-(3,4-Dihydroxyphenyl)-(2S)-2-((2E)-3-(m-tolyl)acrylamido)propanoate (9g)White foam (300 mg, 41.8%); IR (KBr) cm−1: 3352, 1721, 1638; 1H-NMR (DMSO-d6) δ: 2.31 (3H, s), 2.75–2.83 (1H, m), 2.86–2.92 (1H, m), 3.62 (3H, s), 4.45–4.55 (1H, m), 6.44–6.50 (1H, d, J=7.6 Hz, m), 6.60–6.72 (3H, m), 7.14–7.20 (1H, d, J=6.8 Hz), 7.25–7.40 (3H, m), 8.40–8.50 (1H, d, J=7.2 Hz), 8.76 (1H, s); 13C-NMR (DMSO-d6) δ: 20.98, 36.43, 51.85, 54.15, 115.26, 116.18, 119.63, 121.11, 124.63, 127.57, 127.94, 128.66, 130.12, 134.47, 137.91, 139.32, 143.76, 144.78, 164.70, 171.96; MS m/z: 355.14 ([M+H]+).
Methyl (2S)-2-((2E)-3-(3-Chlorophenyl)acrylamido)-3-(3,4-dihydroxyphenyl)propanoate (9h)White foam (400 mg, 52.7%); IR (KBr) cm−1: 3328, 1714, 1659; 1H-NMR (DMSO-d6) δ: 2.75–2.85 (1H, m), 2.86–2.95 (1H, m), 3.62 (3H, s), 4.48–4.60 (1H, m), 6.44–6.50 (1H, d, J=7.6 Hz), 6.56–6.66 (2H, m), 6.72–6.82 (1H, d, J=16.0 Hz), 7.32–7.47 (3H, m), 7.50–7.57 (1H, m), 7.63 (1H, s), 8.36–8.58 (1H, d, J=7.6 Hz), 8.76 (1H, s); 13C-NMR (DMSO-d6) δ: 36.39, 51.86, 54.10, 115.52, 116.16, 119.60, 122.95, 125.86, 127.08, 127.46, 129.01, 130.55, 133.47, 136.83, 137.66, 143.76, 144.77, 164.29, 171.82; MS m/z: 375.09 ([M+H]+).
Methyl 3-(3,4-Dihydroxyphenyl)-(2S)-2-((2E)-3-(3-(trifluoromethyl)phenyl)acrylamido)propanoate (9i)White foam (300 mg, 36.3%); IR (KBr) cm−1: 3353, 1734, 1614; 1H-NMR (DMSO-d6) δ: 2.74–2.82 (1H, m), 2.86–2.92 (1H, m), 3.62 (3H, s), 4.48–4.54 (1H, m), 6.52–6.55 (1H, d, J=8.4 Hz), 6.56–6.64 (2H, m), 6.80–6.88 (1H, d, J=16.8 Hz), 7.44–7.52 (1H, d, J=16.8 Hz), 7.60–7.66 (1H, t, J=8.4 Hz), 7.82–7.88 (1H, d, J=8.4 Hz), 7.88–7.94 (1H, s); 13C-NMR (DMSO-d6) δ: 36.41, 51.93, 54.15, 115.26, 116.17, 119.63, 123.39, 123.82 (d, JC–F=192.6 Hz), 127.45, 129.39 (d, JC–F=31.1 Hz), 129.93, 131.22, 135.73, 137.57, 143.80, 144.80, 164.26, 171.85; MS m/z: 410.20 ([M+H]+).20)
Methyl 3-(3,4-Dihydroxyphenyl)-(2S)-2-((2E)-3-(4-(trifluoromethyl)phenyl)acrylamido)propanoate (9j)White foam (400 mg, 48.4%); IR (KBr) cm−1: 3342, 1734, 1682; 1H-NMR (CDCl3+DMSO-d6) δ: 3.02–2.92 (m, 2H), 3.63 (s, 3H), 4.90–4.80 (m, 1H), 6.48–6.40 (d, J=8.0 Hz, 1H), 6.61 (s, 1H), 6.70–6.65 (d, J=8.0 Hz, 1H), 7.15–7.05 (m, 1H), 7.55–7.45 (m, 5H); 13C-NMR (CDCl3+DMSO-d6) δ: 36.69, 51.77, 53.40, 114.62, 115.55, 120.40, 125.04 (d, JC–F=218.5 Hz), 127.23(d, JC–F=22.7 Hz), 127.45, 137.83, 138.80, 143.06, 143.92, 164.61, 171.36. MS m/z: 410.10 ([M+H]+).
Methyl (2S)-2-((2E)-3-(3,5-Bis(trifluoromethyl)phenyl)acrylamido)-3-(3,4-dihydroxy-phenyl)propanoate (9k)White foam (200 mg, 20.8%); IR (KBr) cm−1: 3348, 1734, 1614; 1H-NMR (DMSO-d6) δ: 2.76–2.82 (1H, m), 2.87–2.96 (1H, m), 3.62 (3H, s), 4.50–4.58 (1H, m), 6.42–6.48 (1H, d, J=7.2 Hz), 6.56–6.64 (2H, m), 6.97–7.04 (1H, d, J=16.4 Hz), 7.54–7.62 (1H, d, J=16.4 Hz), 8.04 (1H, s), 8.24 (2H, s); 13C-NMR (DMSO-d6) δ: 36.43, 51.96, 54.15, 115.32, 116.20, 119.67, 121.70 (d, JC–F=270.7 Hz), 122.29, 125.54, 127.41, 127.82, 130.55 (q, JC–F=32.7 Hz), 136.04, 137.53, 143.84, 144.84, 163.97, 171.73; MS m/z: 478.20 ([M+H]+).
Methyl (2S)-2-((2E)-3-(3,5-Bis(trifluoromethyl)phenyl)propanamido)-3-(3,4-dihydroxyphenyl)propanoate (9l)White foam (500 mg, 51.7%); IR (KBr) cm−1: 3402, 1740, 1652; 1H-NMR (CDCl3+DMSO-d6) δ: 2.62–2.68 (2H, t, J=7.6 Hz), 2.86–2.96 (2H, m), 3.04–3.14 (2H, t, J=7.2 Hz), 3.66 (3H, s), 4.70–4.80 (1H, m), 6.36–6.42 (1H, d, J=8.0 Hz), 6.62 (1H, s), 6.65–6.73 (1H, d, J=8.0 Hz), 7.18 (1H, s), 7.56 (2H, s), 7.72–7.78 (2H, m); 13C-NMR (CDCl3+DMSO-d6) δ: 30.42, 36.13, 36.40, 51.12, 52.90, 119.08, 119.93, 121.29 (d, JC–F=269.2 Hz), 127.04, 128.11, 130.23 (d, JC–F=33.4 Hz), 142.81, 143.21, 143.72, 170.41, 170.92; MS m/z: 480.15 ([M+H]+).
Immortalized Microglia CultureThe BV2 cell (a mouse microglial cell line) developed by Dr. V. Bocchini (University of Perugia, Perugia, Italy) was generously provided by Dr. K. Suk (Kyungpook National University, Daegu, Korea). The immortalized murine BV2 cell line that exhibits both the phenotypic and functional properties of the reactive microglial cells was grown and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% heat-inactivated fetal bovine serum (FBS), at 37°C in a humidified incubator under 5% CO2.
Nitrite QuantificationThe BV2 cells (5×104 cells/mL) in 24-well plates in 500 µL culture medium were pretreated with different concentrations of the synthesized compounds for 30 min after being stimulated with LPS (100 ng/mL) for 24 h. In brief, 50 µL of culture supernatant reacted with an equal volume of Griess reagent (part 0.1% naphthylethylenediamine and part 1% sulfanilamide in 5% H3PO4) in 96-well plates for 10 min at room temperature in the dark. Nitrite concentrations were determined by using standard solutions of sodium nitrite prepared in cell-culture medium. The absorbance at 540 nm was determined using a microplate reader (Molecular Device, U.S.A.). Each experiment was performed in triplicate.20)
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Cytotoxicity AssayThe cell viability of the cultured cells was determined by measuring the reduction of MTT to formazan. Briefly, cells were seeded and treated with different concentrations of the synthesized compounds. After incubation with LPS (100 ng/mL) for 24 h, 0.5 mg/mL of MTT solution was added to each well. After incubation for 2 h at 37°C and 5% CO2, the supernatants were removed and the formed formazan crystals in the viable cells were dissolved in DMSO. The absorbance at 550 nm was determined using a microplate reader (Molecular Device).20)
Immunoblot AnalysisTo obtain the total cell lysate, 100 µL (or 50 µL) of radio immunoprecipitation assay (RIPA) buffer [1×phosphate-buffered saline (PBS), 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), with freshly added protease inhibitor cocktail (Calbiocam, CA, U.S.A.)] was added to the BV2 cells cultured in 6-well plates. The cells were scraped, incubated for 10 min on ice and centrifuged at 14000×rpm for 10 min at 4°C. The protein concentration was determined by the detergent compatible (DC) protein assay from Bio-Rad (Hercules, CA, U.S.A.), and 20 µg of whole cell lysate was separated in 10% SDS-polyacrylamide gels (PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, MA, U.S.A.) using an electroblotting apparatus (Biorad, CA, U.S.A.). The membranes were blocked for 1 h in TBS containing 0.1% Tween-20 and 5% dry milk, and were then incubated overnight with primary antibodies anti-iNOS, 1 : 5000 (Upstate, NY, U.S.A.) followed by incubation for 1 h with horseradish peroxidase-conjugated secondary antibodies (1 : 10000) (Cell signaling, MA, U.S.A.). The optical densities of the antibody-specific bands were analyzed by a Luminescent Image Analyzer, LAS-3000 (Fuji, Japan).20)
Isolation of Total RNA and RT-PCR AnalysisBV2 cells (5×104 cells/mL) were cultured in 6-well plates, and the total RNA was isolated by extraction with TRIzol kit (Invitrogen, CA, U.S.A.). For the RT-PCR, 2.5 µg of total RNA from BV2 cell was used for RT-PCR by using a First Strand cDNA Synthesis kit (Invitrogen). PCR was performed using the above-prepared cDNA as a template. The PCR conditions were as follows: iNOS, 27 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 19 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s. GAPDH was used as an internal control to evaluate the relative expressions of iNOS.20)
Since the inhibitory effect on NO production of clovamide methyl ester 9a was higher than that of clovamide 2 during the preliminary study, all clovamide analogues possessing the methyl ester group in L-dopa moiety were synthesized. Two analogues (7a, b), where the hydroxyl group in either caffeic acid moiety or L-dopa moiety was replaced by a fluorine group, were synthesized as depicted in Chart 1. After carboxylic acids in L-dopa 3 and compound 4 were protected with methanol using thionyl chloride, the resulting two methyl esters (5a, b) were reacted with corresponding cinnamic acid in presence of EDC, 1-hydroxy-benzotriazole (HOBt) and DIEA to produce crude amide products, which were purified by chromatography on silica gel with ethyl acetate–hexane (1 : 1, v/v) to give pure products.

Reagents and conditions: (a) SOCl2, MeOH, reflux, 8 h; (b) i) 4 N HCl in dioxane, r.t., 4 h; ii) SOCl2, MeOH, reflux, 4 h; (c) EDC, HOBt, DIEA, DCM–DMF (4 : 1), r.t., 4 h.
Other clovamide methyl ester analogues (9a–l) were synthesized from L-dopa methyl ester hydrochloride 5a by following Chart 2. L-Dopa methyl ester hydrochloride 5a was reacted with 3-hydroxy cinnamic acid or coumaric acid (8b, c) to afford the crude products of the corresponding amides (9b, c). Similarly, clovamide methyl ester analogues with various substituents such as fluorinated or trifluoromethylated (9d–l) were synthesized from L-dopa methyl ester hydrochloride 5a by reacting with various cinnamic acids or hydrocinnamic acid (8d–l) in the presence of EDC, HOBt, and DIEA to produce the crude products of the corresponding amides (9d–l). The crude products were purified by chromatography on silica gel with ethyl acetate–hexane (1 : 1, v/v) to give pure products.
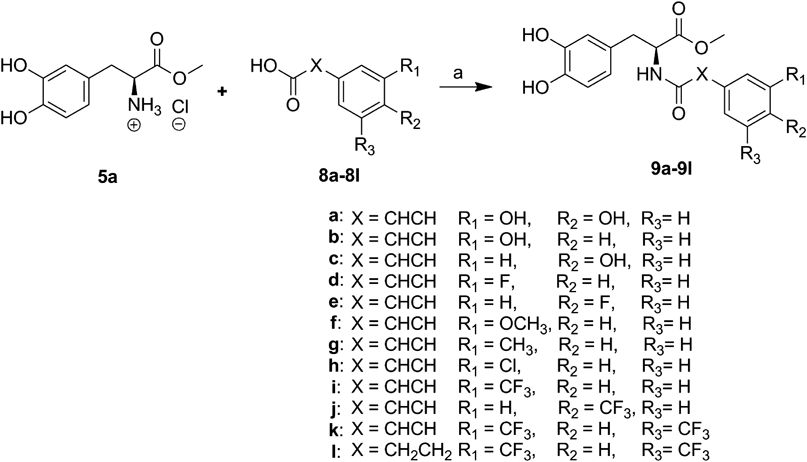
Reagents and conditions: (a) EDC, HOBt, DIEA, DCM–DMF (4 : 1), r.t., 4 h.
Neuroinflammation resulting from the activation of microglia plays an important role in the pathology of neurodegenerative diseases.21) Over-activation of microglia initiates an inflammatory cascade in the central nervous system, propagates the levels of inflammatory and oxidative stress molecules and can lead to neuronal dysfunction and neuronal cell death.22) Among the many inflammatory molecules produced by activated microglia, NO production has been widely used as a representative measure of inflammatory activation of microglia.23) Therefore, in this paper, the inhibitory effect on the production of NO in LPS-induced BV2 cells was evaluated for rosmarinic acid 1, clovamide 2, clovamide methyl ester 9a and its analogues (7a, b, 9b–l). After BV2 cells were co-treated with compounds at a concentration of 10 µM and LPS (100 ng/mL) for 24 h, the nitrite concentration was determined in the supernatant by the Griess assay, and their results are presented in Table 1.
 | |||||
|---|---|---|---|---|---|
| Compounds | X | Substituent (R1) | Substituent (R2) | Inhibition of NO release (%) at 10 µM | IC50 (µM) |
| 1 | 13.2±0.8 | ||||
| 2 | 15.2±3.3 | ||||
| 7a | CH=CH | 3,4-diOH | 3,4-diF | 32.7±0.9 | |
| 7b | CH=CH | 3,4-diF | 3,4-diOH | 10.3±3.4 | |
| 9a | CH=CH | 3,4-diOH | 3,4-diOH | 27.1±2.8 | 73.6 |
| 9b | CH=CH | 3,4-diOH | 3-OH | 23.6±1.0 | |
| 9c | CH=CH | 3,4-diOH | 4-OH | 21.4±0.5 | |
| 9d | CH=CH | 3,4-diOH | 3-F | 49.8±0.5 | |
| 9e | CH=CH | 3,4-diOH | 4-F | 15.6±2.3 | |
| 9f | CH=CH | 3,4-diOH | 3-OCH3 | 19.4±2.3 | |
| 9g | CH=CH | 3,4-diOH | 3-CH3 | 34.1±5.3 | |
| 9h | CH=CH | 3,4-diOH | 3-Cl | 24.1±2.2 | |
| 9i | CH=CH | 3,4-diOH | 3-CF3 | 64.3±1.0 | |
| 9j | CH=CH | 3,4-diOH | 4-CF3 | 20.3±3.4 | |
| 9k | CH=CH | 3,4-diOH | 3,5-diCF3 | 78.0±1.6 | 4.8 |
| 9l | CH2CH2 | 3,4-diOH | 3,5-diCF3 | 97.4±1.1 | 2.8 |
The fluorine substitution, as a bioisostere, for hydrogen or hydroxy group in compounds is a well-known method to change reactivity, metabolic stability, acidity, and membrane permeability of the molecules. Introduction of fluorine, due to resistance of the C–F bond and electronegativity difference between the two atoms, increases the lipophilicity and suppresses the metabolic detoxification by increasing the in vivo lifetime.24,25) For these reasons, fluorine has played important role in drug design and fluorinated drugs have been a popular topic in the field of pharmaceutical science.26,27) Thus, we first investigated the importance of the catechol moiety in both the L-dopa moiety and caffeic acid moiety on activity by replacing the hydroxyl group with a fluorine group. The biological activities of 3,4-difluorophenyl derivative 7a, where the catechol moiety in caffeic acid moiety was modified, and 3,4-difluorophenyl derivative 7b, where the catechol moiety in L-dopa moiety was modified, were tested. The inhibitory effect of 7a was about 3-fold more potent than that of 7b with 32.7±0.9 and 10.3±3.4% at a concentration of 10 µM, respectively. This result indicated that the catechol moiety in caffeic acid can be replaced with another group while substitution of the catechol moiety in L-dopa could lead to loss of activity. Therefore, further modification was focused on the catechol moiety in caffeic acid.
In order to confirm whether both hydroxyl groups in the catechol moiety are essential for the activity, we tested 3-hydroxyphenyl analogue 9b and 4-hydroxyphenyl analogue 9c where one hydroxyl group in catechol moiety in 9a was removed. Both compounds revealed lower activities (23.6±1.0, 21.4±0.5%, respectively) than the parent compound 9a (27.1±2.8%), which indicated that the catechol moiety in 9a is important for retaining activity. However, when we replaced the hydroxyl group in 9b and c with a fluorine group, surprisingly, 3-fluorophenyl analogue 9d revealed higher activity with 49.8±0.5% inhibition than 3-hydroxyphenyl analogue 9b (23.6±1.0%) while 4-fluorophenyl analogue 9e (15.6±2.3%) was less active than the parent compound. This result indicated that the proper group at the meta position of the catechol moiety could enhance the inhibitory effect on NO production. Therefore, we further evaluated the substitution effect of fluorine group in 9d on the NO inhibitory activity by replacing it with bioisosteric groups. The inhibitory effect of 3-methoxyphenyl analogue 9f (19.4±2.3%) and 3-methylphenyl analogue 9g (34.1±5.3%) was less active than that of 9d (49.8±0.5%). In addition, the inhibition of 3-chlorophenyl analogue 9h, where the chlorine group was introduced in meta position, on NO production was reduced to 24.1±2.2%. These results showed that electron withdrawing effect and size of halogen group are important for retaining the activity in this series.
On the basis of our result that fluorine substitution at meta position of catechol moiety enhanced the activity, and previous reports that CF3-bearing molecules could lead to different reactivity and biological properties due to different metabolic consequences,28,29) we then investigated the effect of trifluoromethyl substitution in catechol moiety on the NO inhibitory activity. Inhibition of 3-trifluroromethylphenyl analogue 9i and 4-trifluroromethylphenyl analogue 9j on NO production in LPS-treated BV2 cells were 64.3±1.0, 20.3±3.4%, respectively, which clearly showed that a trifluoromethyl group is more favorable than the comparable fluorine group for enhancing activity. As seen with results of fluorophenyl analogues 9d and e, a similar pattern was also reproduced with trifluoromethyl substitution since the NO inhibition of 3-trifluoromethylphenyl analogue 9i is more active than 4-trifluoromethylphenyl analogue 9j. The enhanced activity of 9i where substitution of a trifluoromethylphenyl ring at meta-position prompted us to evaluate 3,5-ditriflurormethylphenyl analogue 9k where an additional trifluoromethyl group is introduced at meta position of phenyl ring. As expected, 9k further improved activity (78.0±1.6%), with IC50 value of 4.8 µM.
Although caffeic acid and its reduced form, dihydrocaffeic acid, generally did not show significant difference in liver lipid peroxidation,30) we tested the importance of the double bond on the activity by preparing analogue 9l, which possessed two trifluoromethyl groups at 3,5-position in catechol moiety but is devoid of a double bond. Surprisingly, the saturated analogue 9l exhibited significantly improved inhibition (97.4±1.1%) with IC50 value of 2.8 µM, which implied that the saturated form in caffeic acid moiety is more favorable for activity in this series. From the result of structure–activity relationship, it was found that additional introduction of trifluoromethyl group at the meta position of the catechol moiety ring and reduction of the double bond in the analogue 9i significantly enhance the inhibition of NO production. Analogue 9l also exhibited a NO inhibitory activity in a dose-dependent manner (Fig. 2).
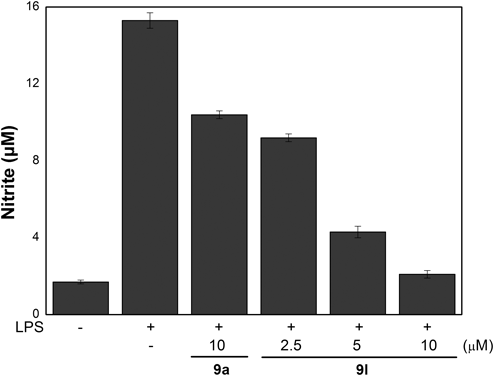
BV2 cells were stimulated with LPS (100 ng/mL) or co-treated with LPS (100 ng/mL) and 9l at concentrations of 2.5, 5.0 and 10 µM for 24 h. The amounts of NO were determined using the Griess reagent. All the data of each group are expressed as the mean±S.E.M. and are representative of results obtained from three independent experiments.
To determine whether the inhibitory effects of clovamide methyl ester (9a) and its analogues (7a, b, 9b–l) on NO production were related to their toxicity, the cytotoxicities of clovamide methyl ester’s analogues were evaluated by MTT assay.31) As shown in Fig. 3, there was no suppression in the cell viabilities of the BV2 cells after being treated with clovamide methyl ester and its analogues at the concentrations of 10 µM. This result confirmed that the inhibitory effects on NO production were due solely to its intrinsic activities rather than its cytotoxicity.

Viability was evaluated using the MTT assay. Cells were incubated with LPS (100 ng/mL) and each sample (clovamide methyl ester and its analogues) for 24 h. The results are displayed as a percentage of the control samples. Each value indicates the mean±S.E.M. and is representative of the results obtained from three independent experiments.
NO produced from ι-arginine by NOS is present in three different types of isozymes, such as endothelial NO (eNOS), neuronal NO (nNOS) and inducible NO (iNOS).32) eNOS and nNOS produce a small amount of NO whereas iNOS produces a large amount of NO, which causes various diseases such as stroke, Alzheimer’s disease, Parkinson’s disease and septic shock.33) Since the reduced NO production is mainly due to the inhibited iNOS expression, we examined whether the NO production was caused by a decrease in the mRNA level of iNOS. LPS-stimulated BV2 cells were treated with 9l and then the amount of iNOS mRNA was measured by RT-PCR. The expression of iNOS mRNA in LPS-treated BV2 cells was significantly increased in comparison with the non-treated control cells. When the LPS-treated BV2 cells were treated with 9l at different concentrations (2.5, 5, 10 µM), the densities of iNOS mRNA in the cells were decreased (Fig. 4(A)). To confirm the effects of 9l on iNOS protein expression, the amount of iNOS protein expression was measured by the immunoblotting method. As shown in Fig. 4(B), the unstimulated cells released basal levels of iNOS protein, while a significant increase of iNOS protein was detected in LPS-stimulated BV-2 cells. When the LPS-treated BV2 cells were treated with 9l at different concentrations (2.5, 5, 10 µM), the densities of iNOS protein in the cells were decreased in a dose-dependent manner. These results demonstrated that the inhibitory action of 9l on NO production was related to a modulation of iNOS induction.
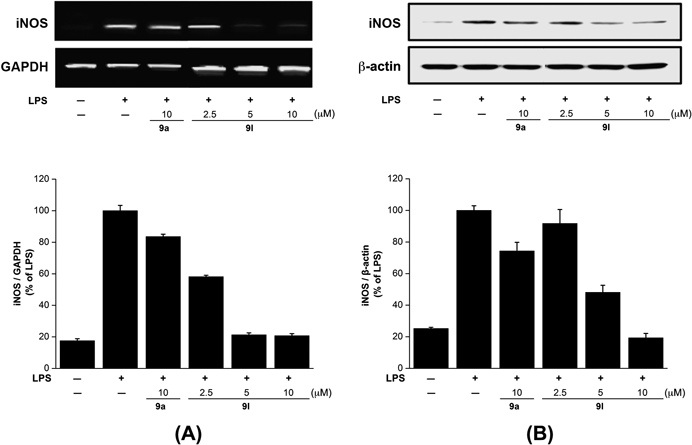
BV2 microglial cells were pretreated with the indicated concentrations of 9l for 30 min before incubating with LPS (100 ng/mL) for 18 h. The cells were lysed, and the lysates were prepared for mRNA levels (A) and analyzed by immunoblotting with an anti-iNOS antibody (B).
We synthesized various clovamide methyl ester analogues where the dihydroxyl group in the catechol moiety were replaced with various groups, and tested their anti-inflammatory activities. Among these synthesized clovamide analogues, the fluorinated analogues (9d, i, k, l) bearing the F or CF3 group at the meta position of the catechol moiety significantly improved the inhibitory effect on NO production in LPS-treated BV2 compared to the parent analogue 9a. In particular, 9l, which has two trifluoromethyl groups at the meta position of the catechol moiety ring and is devoid of double bond, inhibited not only NO production 26.3 times better than that of the clovamide methyl ester 9a, but also iNOS expression in LPS-induced BV2 cells without cytotoxicity. These preliminary structure–activity relationship results of clovamide broaden our understanding of the pharmacophore of clovamide, and 9l can be used as a possible new anti-inflammatory agent.
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) Grant funded by the Ministry of Education [NRF-2014R1A1A2057316 (to S.-H. Yoon) and (NRF-2012R1A1A2042207 (to J.-Y. Park)].
The authors declare no conflict of interest.