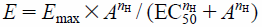Abstract
The present study aimed to investigate the potential inhibitory effects of 21 clinically available hypnotics on acetylcholine (ACh)-induced contractions in rat urinary bladder smooth muscle (UBSM) in order to predict whether these hypnotics could induce voiding impairment. ACh-induced contraction in rat UBSM was inhibited only by diphenhydramine (a histamine H1 receptor antagonist) at a concentration that was clinically relevant. ACh-induced contraction was also significantly inhibited by flurazepam (a benzodiazepine hypnotic) and suvorexant (an orexin receptor antagonist), albeit at concentrations that substantially exceeded clinically achievable blood levels. These three drugs (at 10−5 M) also inhibited high-KCl (80 mM) Locke–Ringer solution-induced contractions. In contrast to the effects of the abovementioned hypnotics, ACh-induced contractions were not significantly affected by triazolam, etizolam, brotizolam, lormetazepam, estazolam, flunitrazepam, nitrazepam (benzodiazepine hypnotics), thiopental, thiamylal, pentobarbital, amobarbital, secobarbital, phenobarbital (barbiturate hypnotics), zolpidem (an imidazopyridine hypnotic), zopiclone (a cyclopyrrolone hypnotic), ramelteon (a melatonin receptor agonist), bromovalerylurea, and chloral hydrate. These findings suggest that most clinically used hypnotics are not likely to result in anticholinergic-induced dysuria within their clinically achievable blood concentration ranges. Diphenhydramine may, however, induce voiding impairment, an action attributable to diminished UBSM contractility within its clinical dose range.
INTRODUCTION
Insomnia is defined as sleep disorder in which any of three symptoms (i.e., difficulty falling asleep, nocturnal awakening, or early morning awakening) are observed. These symptoms occur repeatedly, despite adequate sleep time and opportunity, resulting in daytime adverse effects.1) Pharmacotherapy using hypnotics is currently used to combat insomnia in patients.2) Insomnia has also been reported as a contributing factor in the development of numerous mental disorders, particularly depression and anxiety disorders.3,4) As the number of patients suffering from mental disorders, such as depression, is increasing worldwide,5) the use of hypnotics as a pharmacotherapy is assumed to be increasing correspondingly. Indeed, in the United States, the number and percentage of office visits for sleep-related problems and those resulting in a prescription for nonbenzodiazepine sleep medications (ca. 350%), benzodiazepine receptor agonists (ca. 430%), and any sleep medication (ca. 200%) have been strikingly increasing from 1999–2010.6)
In their brief history, many hypnotics have been chemically synthesized and used clinically. The first forms of hypnotics were halogen compounds, such as chloral hydrate, and bromide hypnotics, such as bromovalerylurea. These were followed by barbiturate hypnotics. However, the abovementioned hypnotics demonstrated a strong dependency and were prone to toxic adverse effects. In the 1960s, benzodiazepine hypnotics, which lack these problems, were introduced. Hypnotics belonging to this category became the first pharmacotherapy choice for insomnia, which is still true today.2,7–9) In the 1980s, zopiclone (a cyclopyrrolone hypnotic) and zolpidem (an imidazopyridine hypnotic), which bind to benzodiazepine binding sites on the type A receptor for gamma-aminobutyric acid (GABAA receptor) despite having different chemical structures from the benzodiazepine derivatives, began to be used clinically.10) After 2000, new hypnotics, such as the melatonin receptor agonist ramelteon and the orexin receptor antagonist suvorexant, both of which do not act on the GABAA receptor, were established.2) Clinical applications of many types of hypnotics have allowed patients with insomnia to select a therapeutic agent that is most suitable for their specific symptoms.
Even with a wide variety of choices, pharmacotherapy using benzodiazepine hypnotics remains the main treatment for insomnia. Benzodiazepine drugs generally exhibit anticholinergic effects and are mostly contraindicated for patients with acute narrow-angle glaucoma as their anticholinergic activities may raise intraocular pressures.11) If these hypnotics were to exert anticholinergic effects on urinary bladder (UB) smooth muscle (UBSM), the UBSM contractile force should be attenuated, resulting in dysuria in patients with benign prostatic hyperplasia (BPH). More importantly, when drug-induced dysuria, resulting in urinary retention, occurs, there is an increased risk of medication interruption, leading to reduced patient adherence to treatment. Despite these potential effects on the function of the lower urinary tract, little information is currently available on the extent of the inhibitory effects of hypnotics on UB contractile function; few systematic pharmacological studies have examined the inhibitory effects of every clinically available hypnotic medication on UB contractions.
Thus, the present study was performed to determine the extent of the inhibition exerted by all hypnotics, which are clinically indicated for insomnia in Japan, on UB contractile function. Since this study aimed to determine the extent of anticholinergic action-derived UB contractile dysfunction as a side effect of insomnia therapy, we focused on the contractions of isolated UBSM preparations in response to acetylcholine (ACh) and investigated the possible inhibitory effects of 21 hypnotics on these contractile responses.
MATERIALS AND METHODS
DrugsThe 21 hypnotics tested in this study were as follows: phenobarbital (Daiichi Sankyo Co., Ltd., Tokyo, Japan); amobarbital and thiopental sodium (Nippon Shinyaku Co., Ltd., Kyoto, Japan); secobarbital sodium and thiamylal sodium (Nichi-Iko Pharmaceutical Co., Ltd., Toyama, Japan); flunitrazepam (Chugai Pharmaceutical Co., Ltd., Tokyo, Japan); diphenhydramine hydrochloride, triazolam, and zolpidem (Sigma-Aldrich Co., St. Louis, MO, U.S.A.); brotizolam, estazolam, flurazepam, lormetazepam, and nitrazepam (FUJIFILM Wako Pure Chemical Industries, Ltd., Osaka, Japan); 1-(2-bromoisovaleryl)urea (bromovalerylurea), chloral hydrate, etizolam, pentobarbital sodium salt, and zopiclone (Tokyo Chemical Industry Co., Ltd., Tokyo, Japan); ramelteon (ChemScene LLC, Monmouth Junction, NJ, U.S.A.); and suvorexant (AdooQ BioScience LLC, Irvine, CA, U.S.A.). ACh chloride was purchased from Daiichi Sankyo Co., Ltd. Atropine sulfate, indomethacin, prazosin hydrochloride, propranolol hydrochloride, and (±)-verapamil were purchased from Sigma-Aldrich Co. [N-Methyl-3H]scopolamine methyl chloride ([3H]NMS, 2960 GBq/mmol) was purchased from PerkinElmer, Inc. Life Sciences, Inc. (Boston, MA, U.S.A.). All other chemicals used were commercially available and of reagent grade.
Amobarbital, bromovalerylurea, etizolam, estazolam, flurazepam, lormetazepam, phenobarbital, ramelteon, and zolpidem were dissolved in ethanol as stock solutions of 2 × 10−2 M, and diluted further with distilled water to the desired concentrations. Brotizolam and zopiclone were dissolved in ethanol as stock solutions of 10−2 M, and diluted further with distilled water to the desired concentrations. Flunitrazepam was dissolved in ethanol as a stock solution of 2 × 10−3 M and diluted further with distilled water to the desired concentrations. Nitrazepam and triazolam were dissolved in dimethyl sulfoxide (DMSO) as stock solutions of 2 × 10−2 and 10−2 M, respectively, and diluted further with distilled water to the desired concentrations. Suvorexant was dissolved in DMSO as a stock solution of 2 × 10−3 M and diluted further with distilled water to the desired concentrations. All other drugs were prepared as aqueous stock solutions and were diluted with distilled water. In the radioligand binding assays, the tested hypnotics (i.e., flurazepam, suvorexant, and diphenhydramine) and atropine were dissolved in DMSO as a stock solution of 2 × 10−2 M and diluted further with DMSO to the desired concentrations.
AnimalsMale Wistar rats (8–9 weeks old; weight 180–250 g, Sankyo Labo Service Corporation, Tokyo, Japan) and male ddY mice (8–10 weeks old; weight 35–47 g, Sankyo Labo Service Corporation) were housed under controlled conditions (21–22°C, relative air humidity 50 ± 5%), and a fixed 12 h light–dark cycle (08:00–20:00) with food and water available ad libitum. This study was approved by the Toho University Animal Care and User Committee (approval number: 15-51-294, accredited on May 22, 2015; approval number: 16-52-294, accredited on May 16, 2016; approval number: 17-53-294, accredited on May 17, 2017; approval number: 18-54-294, accredited on May 7, 2018) and was conducted in accordance with the User’s Guideline to the Laboratory Animal Center of Faculty of Pharmaceutical Sciences, Toho University.
UB PreparationsThe rats were anesthetized with isoflurane (inhalation) and exsanguinated from a carotid artery. The UB was immediately removed and placed in Locke–Ringer solution of the following composition (mM): NaCl (154), KCl (5.6), CaCl2 (2.2), MgCl2 (2.1), NaHCO3 (5.9), and glucose (2.8). The isolated UB, after removing the surrounding adipose tissue, connective tissue, and bladder trigone, was opened with a longitudinal incision and UB strips (approximately 2 mm in width × 25 mm in length) were prepared in Locke–Ringer solution. The UB strips were suspended in a 20-mL organ bath containing Locke–Ringer solution, which was aerated with 95% O2 and 5% CO2 and maintained at 32 ± 1°C. The strips were subjected to a tension of 0.5 g and allowed to equilibrate for 20 min. Changes in the mechanical activity of the strips were recorded isotonically. All experiments were carried out in the presence of indomethacin (3 × 10−6 M) to block any influence of endogenous prostaglandins.
Assessment of Effects of Hypnotics on ACh-Induced UBSM ContractionFirst, the UBSM preparation was contracted using 10−4 M ACh at least three times at 20 min intervals. After a 30-min equilibration period, ACh (10−7–3 × 10−3 M) was incrementally applied to the bath medium until a maximum response was obtained. This procedure was repeated, and this second ACh concentration-response curve was considered the control response. Following this procedure, to assess the effect of each hypnotic (3 × 10−7–10−5 M), atropine (3 × 10−9–10−7 M), or verapamil (10−5 M), on ACh-induced UBSM contraction, ACh concentration-response curves were obtained in the manner previously described in the presence of different concentrations of each tested drug. The UBSM preparation was preincubated with the hypnotic, atropine, or verapamil for 30 min prior to the cumulative application of ACh.
Assessment of Effects of Hypnotics on UBSM Contraction Induced by High-KCl Locke–Ringer SolutionThe UBSM preparation was first contracted using 10−4 M ACh at least three times at 20-min intervals. When the tension recovered to the level before ACh administration, atropine (10−6 M), prazosin (3 × 10−7 M), and propranolol (10−7 M) were added to the bath medium to prevent any potential effects on muscarinic receptors, α1-adrenoceptors, and β-adrenoceptors, respectively. After a 30-min equilibration period, to produce sustained contractions, the strip was contracted with high-KCl Locke–Ringer solution (containing atropine, prazosin, and propranolol, as indicated above) of the following composition (mM): NaCl (79.6), KCl (80), CaCl2 (2.2), MgCl2 (2.1), NaHCO3 (5.9), and glucose (2.8). When the contractile response reached steady-state, the tested hypnotic (i.e., flurazepam, suvorexant, or diphenhydramine; 10−7–10−5 M) was incrementally applied to the bath medium. To confirm a maximal inhibitory response at the end of the experiment, the USBM preparation was treated with verapamil (10−5 M). The inhibitory effects of the hypnotics on sustained UBSM contractions were expressed as percent relaxation. The values were calculated considering the tension just before administration of hypnotics to be 100% contraction, and the basal tension before application of high-KCl Locke–Ringer solution to be 0% contraction.
Muscarinic Receptor Binding AssayThe radioligand binding assays for muscarinic receptors were performed using the antagonistic radioligand, [3H]NMS, as previously described by Harada et al.12) Briefly, the mouse cerebral cortex tissues were homogenized using a Polytron® homogenizer (Kinematica AG, Lucerne, Switzerland) in ice-cold 30 mM Na+/N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) buffer (pH 7.5). The homogenates were then centrifuged at 49000 × g for 20 min at 4°C in an ultracentrifuge (himac CP-NX series; CP 80NX, Koki Holdings Co., Ltd., Tokyo, Japan). The resulting pellets were resuspended in 19 volumes of ice-cold HEPES buffer. These homogenates were further diluted 5-fold with ice-cold HEPES buffer immediately before use. In the saturation experiments, mouse tissue homogenates were incubated with increasing concentrations (0.0625–2 nM) of [3H]NMS.
In the displacement experiments, mouse cerebral cortex tissue homogenates were incubated with [3H]NMS (0.5 nM) in the presence of various concentrations of the tested hypnotics (i.e., flurazepam, suvorexant, or diphenhydramine). In this experiment, the concentration of DMSO in the mixtures was adjusted to be 0.5%. The mixtures were incubated at 25°C for 60 min, the reaction was terminated by rapid filtration through Whatman GF/B glass fiber filters, and the filters were rinsed with ice-cold HEPES buffer. The filters were immersed in scintillation fluid and radioactivity was determined by liquid scintillation counting. Specific binding for [3H]NMS was determined experimentally as the difference between counts in the absence and presence of 1 µM atropine. This series of experiments were performed in duplicate.
Data Analysis and StatisticsTo construct concentration-response curves for ACh-induced contractions, the tension level before cumulative application of ACh was defined as 0% contraction, and the maximum contraction obtained with 3 × 10−3 M ACh in the control response was designated as 100%. The data were plotted as a function of ACh concentration and fitted to the equation:
where E is the % contraction at a given concentration, Emax is the maximum response, A is the ACh concentration, nH is Hill coefficient, and EC50 is the ACh concentration producing a 50% response. Curve-fitting was performed using GraphPad Prism™ (Version 6.07; GraphPad Software, Inc., San Diego, CA, U.S.A.).
When possible, the competitive antagonistic potency of the tested hypnotics was expressed as a pA2 value and calculated from a Schild plot analysis of hypnotics versus ACh concentrations by using concentration-response curves for the ACh-induced contractions.13)
Binding data were subjected to nonlinear regression analyses using GraphPad Prism™ as previously described by Harada et al.12) The dissociation constant (Kd) and maximal number of binding sites (Bmax) for [3H]NMS were determined by Scatchard analysis for the specific binding of [3H]NMS. The abilities of the tested hypnotics to inhibit specific binding of the radioligand ([3H]NMS) were estimated from their IC50 values (i.e., the molar concentration of the unlabeled drug necessary to displace 50% of the specific binding of [3H]NMS). The inhibition constant (Ki) was calculated from the equation Ki = IC50/(1 + L/Kd), where L is the concentration of [3H]NMS.
All values in the text and illustrations are presented as means ± standard error of the mean (S.E.M.) or mean values with 95% confidence intervals (CIs) of the data obtained from different numbers (n) of preparations. GraphPad Prism™ was used for statistical analysis. Differences among concentration-response curves were evaluated using two-way ANOVA followed by Dunnett’s multiple comparison post-tests. p values less than 0.05 were considered to indicate statistically significant differences.
RESULTS
Effects of Atropine on ACh-Induced UBSM ContractionsFigure 1 shows the effect of atropine (3 × 10−9–10−7 M) on ACh-induced contraction in rat UBSM. Atropine (3 × 10−9–10−7 M) shifted the concentration response curves for ACh rightward in a concentration-dependent manner (Fig. 1A). The inhibitory effects of atropine on ACh-induced contractions were deemed to be based on competitive antagonism, since the slope of the regression line for the Schild plot of atropine vs. ACh was 0.89 (95% CI: 0.69–1.10), which was not different from unity (Fig. 1B). The pA2 value of atropine was calculated as 8.81 (95% CI: 8.50–9.23). Based on the above results, we determined that the anticholinergic effects of various hypnotics can be evaluated using their inhibitory effects on ACh-induced contraction in rat UBSM as indexes.
Effects of Benzodiazepine Hypnotics on ACh-Induced UBSM ContractionsFigure 2 shows the effects of benzodiazepine hypnotics on the concentration-response curves of ACh. Of the tested drugs, triazolam (Fig. 2A), etizolam (Fig. 2B), brotizolam (Fig. 2C), lormetazepam (Fig. 2D), estazolam (Fig. 2E), flunitrazepam (Fig. 2F), and nitrazepam (Fig. 2G) did not significantly affect ACh-induced contraction in the concentration range of 3 × 10−7–10−5 M.
In contrast, flurazepam (Fig. 2H) inhibited ACh-induced contraction in the concentration range of 3 × 10−7–10−5 M, which was determined to be statistically significant using two-way ANOVA to compare the concentration-response curves vs. control (Fig. 2Ha). The slope of the regression line for the Schild plot of flurazepam vs. ACh was 0.67 (95% CI: 0.31–1.03), which was far less than and not significantly different from unity (Fig. 2Hb). Therefore, we could not clearly deduce whether the inhibitory effects of flurazepam were caused by competitive antagonism of ACh, or if other effects mediated this inhibition. If a competitive antagonism was assumed to exist for flurazepam vs. ACh, the pA2 value for flurazepam would be 5.69 (95% CI: 5.30–6.03).
Effects of Barbiturate Hypnotics on ACh-Induced UBSM ContractionsFigure 3 shows the effects of barbiturate hypnotics on the concentration-response curves of ACh. None of the tested drugs (thiopental (Fig. 3A), thiamylal (Fig. 3B), pentobarbital (Fig. 3C), amobarbital (Fig. 3D), secobarbital (Fig. 3E), and phenobarbital (Fig. 3F)) significantly affected ACh-induced contraction in the concentration range of 3 × 10−7–10−5 M.
Effects of Other Hypnotics on ACh-Induced UBSM ContractionsFigure 4 shows the effects of the other hypnotics on the concentration response curves for ACh. Of the tested drugs, zolpidem (an imidazopyridine hypnotic) (Fig. 4A), zopiclone (a cyclopyrrolone hypnotic) (Fig. 4B), ramelteon (a melatonin receptor agonist) (Fig. 4C), bromovalerylurea (Fig. 4E), and chloral hydrate (Fig. 4F) did not significantly affect ACh-induced contraction in the concentration range of 3 × 10−7–10−5 M.
In contrast, suvorexant (Fig. 4D) significantly inhibited ACh-induced contraction in the concentration range of 10−6–10−5 M, which was determined to be statistically significant using two-way ANOVA to compare the concentration-response curves vs. control (Fig. 4Da). However, the slope of the regression line for the Schild plot analysis of suvorexant vs. ACh was 0.20 (95% CI: −0.69–1.08), which was far less than and not significantly different from unity (Fig. 4Db). Therefore, we deemed that the inhibitory effects of flurazepam were not caused by competitive antagonism against ACh.
Diphenhydramine (Fig. 4G) also significantly inhibited ACh-induced contraction in the concentration range of 3 × 10−7–10−5 M, which was determined to be statistically significant using two-way ANOVA to compare the concentration-response curves vs. control (Fig. 4Ga). The slope of the regression line for the Schild plot analysis of diphenhydramine vs. ACh was 0.83 (95% CI: 0.72–0.94), which was very close to, but not significantly different from unity (Fig. 4Gb). We, therefore, inferred that competitive antagonism against ACh may be involved in the inhibitory effects of diphenhydramine on ACh-induced contractions. Assuming that diphenhydramine competitively antagonized ACh, the calculated pA2 value for diphenhydramine would be 6.73 (95% CI: 6.59–6.90).
Effects of Hypnotics on UBSM Contraction Induced by High-KCl Locke–Ringer SolutionFlurazepam, suvorexant, and diphenhydramine significantly inhibited ACh-induced contractions. However, these drugs did not show a clear competitive antagonism against ACh (i.e., the slopes of their regression lines for the Schild plot analyses were not statistically different from unity). As one reason why these hypnotics did not show clear ACh competitive antagonism, we postulated that they possess Ca2+ antagonistic actions. This would then affect the concentration response curves for ACh in a noncompetitive fashion. Thus, we investigated this possibility pharmacologically. First, we investigated the effect of verapamil, a Ca2+ channel inhibitor, on ACh-induced contractions, to determine whether they are mediated by the activation of L-type Ca2+ channels.
Figure 5 shows that the maximum contractile response attained with 3 × 10−3 M ACh was suppressed to 43.6 ± 7.2% (n = 4) by pretreatment with 10−5 M verapamil. This finding suggests that ACh-induced UB contraction is partly mediated via Ca2+ influx through L-type Ca2+ channels. Therefore, we next investigated whether the drugs showing inhibitory effects on ACh-induced contractions (i.e., flurazepam, suvorexant, and diphenhydramine) could suppress the depolarizing contractions induced by high-potassium solution. Specifically, if these drugs had inhibitory effects on L-type Ca2+ channels, they were expected to inhibit high potassium-induced contractions, which were induced by Ca2+ influx through these channels.
Figure 6 shows the effects of flurazepam, suvorexant, and diphenhydramine (10−7–10−5 M) on the depolarizing contractile responses to 80 mM KCl solution. All drugs were found to strongly inhibit high KCl-induced contractions at 10−5 M, with the remaining contractile components following drug treatment as follows: 70.3 ± 6.0% (n = 4) for flurazepam (Fig. 6A), 50.0 ± 7.4% (n = 4) for suvorexant (Fig. 6B), and 26.0 ± 4.3% (n = 4) for diphenhydramine (Fig. 6C). Because the residual contractile component in the presence of each drug was found to be completely suppressed by verapamil (10−5 M) (Figs. 6A–C), the contractions induced by high KCl solution were determined to be dependent on L-type Ca2+ channels.
Effects of Hypnotics on Specific Binding of [3H]NMS in Mouse Cerebral CortexIn order to obtain biochemical evidence to support the speculations deduced by the pharmacological mechanistic studies mentioned above, the inhibitory effects of the three hypnotics (flurazepam, suvorexant, and diphenhydramine) on the specific binding of [3H]NMS were examined. We found that both flurazepam and suvorexant hardly affect [3H]NMS specific binding until 10−5 M (n = 3), although 10−4 M flurazepam and suvorexant inhibited binding by 23.2 ± 1.8% (n = 3) and 42.1 ± 1.1% (n = 3), respectively (Figs. 7A, B). In contrast, diphenhydramine concentration-dependently inhibited [3H]NMS specific binding at 10−8–10−4 M (Fig. 7C). The pKi value of diphenhydramine was calculated to be 5.97 ± 0.04 (n = 3). This value was almost identical to its pA2 value (6.73), which was calculated based on the assumption that diphenhydramine competitively antagonizes ACh.
DISCUSSION
Benzodiazepine hypnotics, which are mainly used as insomnia medications, are generally recognized to have weak anticholinergic activity. Most of these hypnotics are contraindicated for patients with acute narrow-angle glaucoma and some are cautioned, although not contraindicated, for use by patients with BPH. However, to date, little information is available regarding whether benzodiazepine hypnotics exhibit anticholinergic actions that are attributable to inhibitory effects exerted on UB motility. If this is true, we wanted to examine the extent of their inhibitory effects. Therefore, we investigated the effects of benzodiazepine hypnotics on ACh-induced contraction of isolated UB preparations to examine whether these drugs could inhibit the micturition reflex and thus, dysuria. We also examined the effects of other insomnia medications, namely barbiturates, cyclopyrrolones, imidazopyridines, a melatonin receptor agonist, and an orexin receptor antagonist, on ACh-induced UB contractions.
Of the hypnotics examined in this study, most (i.e., triazolam, etizolam, brotizolam, lormetazepam, estazolam, flunitrazepam, nitrazepam (benzodiazepines), thiopental, thiamylal, pentobarbital, amobarbital, secobarbital, phenobarbital (barbiturates), zopiclone (a cyclopyrrolone), zolpidem (an imidazopyridine), ramelteon (a melatonin receptor agonist), bromovalerylurea, and chloral hydrate) did not suppress ACh-induced contraction. Therefore, these hypnotics are not likely to cause anticholinergic-based suppression of UB contraction and dysuria. In contrast, flurazepam (a benzodiazepine), suvorexant (an orexin receptor antagonist), and diphenhydramine (a histamine H1 receptor antagonist) showed a significant inhibitory effect on ACh-induced contraction.
For the hypnotics that showed a significant inhibitory effect on ACh-induced contraction, the slope of the regression line for the Schild plot was 0.67 and 0.20 for flurazepam and suvorexant, respectively. Therefore, a competitive antagonistic action at the ACh receptor (muscarinic receptor) does not seem to explain the inhibition of ACh-induced contraction by these drugs. Our observation that both flurazepam and suvorexant did not inhibit the specific binding of [3H]NMS supports that assertion. In contrast, both flurazepam (10−5 M) and suvorexant (10−5 M) significantly inhibited the contractile responses induced by high-KCl solution. Therefore, the suppression of ACh-induced contraction by flurazepam and suvorexant was not attributed to their competitive antagonistic actions against ACh. Instead, it may result from Ca2+ antagonistic actions (i.e., inhibition of voltage-dependent L-type Ca2+ channels). Indeed, since the ACh-induced contraction of UB preparations was found to be suppressed to 50% or less by verapamil, the L-type Ca2+ channel was suggested to be involved in the generation of the contractile response. Therefore, it is reasonable to speculate that the suppression of ACh-induced contraction by flurazepam and suvorexant is partly mediated by their Ca2+ antagonistic actions. However, their clinically achievable concentrations are reported to be 0.82–1.7 ng/mL (1.93–4.0 nM) for flurazepam,14) and 0.605–0.767 µM for suvorexant.15) These blood concentrations are lower than the concentrations required to inhibit ACh-induced contractions (i.e., 3 × 10−7 and 10−6 M for flurazepam and suvorexant, respectively). Thus, flurazepam and suvorexant are unlikely to exhibit anticholinergic action-based suppression of UB contraction and dysuria, as long as they are used at the recommended clinical dose. However, suvorexant was reported to cause dysuria, abnormal urination, and nocturia (one case each) in a phase III international joint study using higher than the approved dose.15) In Europe, flurazepam was also reported to cause dysuria as a rare side effect.16) A previous report that flurazepam inhibits Ca2+ channels at clinically relevant concentrations17) corroborates the interpretation of our research results.
The possible Ca2+ channel inhibitory actions of flurazepam and suvorexant may be explained by their chemical structural similarities to Ca2+ channel inhibitors. Regarding flurazepam, its chemical structure is similar to that of clentiazem, a benzothiazepine Ca2+ channel inhibitor (Fig. 8); flurazepam contains a benzodiazepine ring, which is very similar in structure to the benzothiazepine ring in clentiazem. Both flurazepam and clentiazem contain a chlorine group and a phenyl group that are bonded to these rings. The side chain structures of these rings are also similar; a diethylaminoethyl group and a dimethylaminoethyl group are bonded to the nitrogen in the flurazepam and clentiazem rings, respectively. Considering suvorexant, structural similarities are seen with zonisamide, which possesses Ca2+ channel inhibitory activity. Particularly, suvorexant has a benzoxazole ring, and this structure is very similar to the benzoisoxazole ring of zonisamide (Fig. 8). Similar ring structures are also found in fostedil (a benzothiazole ring) and mibefradil (a benzimidazole ring) (Fig. 8). Since zonisamide has a simple chemical structure composed of a methanesulfonamide-substituted benzoisoxazole ring, any Ca2+ channel inhibitory actions of suvorexant are likely to be closely associated with its benzoxazole ring.
For diphenhydramine, the slope of the regression line for the Schild plot could be regarded as close to unity; however, the value (0.83) did not exist within the 95% CI. Thus, the inhibition of ACh-induced contraction by diphenhydramine can be partly explained by competitive antagonism of ACh. The calculated pA2 value for diphenhydramine was 6.73 when the slope of the regression line of the Schild plot was considered to be unity. Since this value is consistent with the reported value vs. carbachol in guinea pig trachea (6.3 ± 0.3),18) our interpretation that the inhibitory effects of diphenhydramine include competitive antagonism against ACh may be reasonable. In the present study, the binding experiments also demonstrated that diphenhydramine is a competitive antagonist of the muscarinic ACh receptor, which was strongly supported by the equivalence of the pKi and pA2 values. However, since diphenhydramine at 10−6–10−5 M was found to strongly suppress high KCl-induced contractions, this drug was also shown to possess Ca2+ antagonistic action within this concentration range. Therefore, the inhibitory actions of diphenhydramine against ACh are suggested to be the result of antagonistic action at the ACh M3 receptor and Ca2+ antagonistic action.
The clinical dose of diphenhydramine as a hypnotic is 50 mg/person (single dose), with a maximum obtainable blood concentration of 66.3 ± 6.9 ng/mL (0.23 ± 0.02 µM).19) The negative logarithmic value (−log) of this blood concentration is 6.60–6.68, which is consistent with the pA2 value (6.73) of diphenhydramine vs. ACh. Therefore, diphenhydramine is suggested to exert its anticholinergic action within clinically relevant doses, and consequently, suppress UB contraction and induce dysuria. Indeed, it was reported that dysuria was caused by diphenhydramine in a patient with BPH,20) indicating the appropriateness of our interpretation of our results.
Diphenhydramine was developed as an antiallergic drug, but intense drowsiness is a side effect. However, diphenhydramine has been sold in recent years as a sleep aid at street drugstores, exploiting this side effect as its principal action. Diphenhydramine is also sold as a preventive treatment for motion sickness. All of these can be purchased as OTC drugs. According to a U.S. survey, many elderly people reportedly take diphenhydramine-containing OTC sleep aids without recognizing the risks associated with their side effects.21) Based on the results of the present study, upon the sale of diphenhydramine-containing OTC sleep aids, pharmacists and other healthcare providers are recommended to note whether the patients are free from lower urinary symptoms such as BPH, and to carefully explain diphenhydramine’s side effects, including dysuria.
In summary, most clinically used hypnotics do not exhibit anticholinergic action-mediated suppression of UB contraction and subsequent dysuria. However, inhibitory actions against ACh-induced UB contraction were found to be exerted by flurazepam (a benzodiazepine), suvorexant (an orexin receptor antagonist), and diphenhydramine (a histamine H1 receptor antagonist). More specifically, diphenhydramine was observed to be a hypnotic that can suppress UB contraction and subsequently induce dysuria, since the anticholinergic effects of this drug can be realized within its clinical dose range.
Acknowledgments
This study was partly supported by The Joint Research Grants of Toho University Faculty of Pharmaceutical Sciences (K.O.).
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) American Academy of Sleep Medicine. International Classification of Sleep Disorders, third edition. American Academy of Sleep Medicine, Darien (2014).
- 2) Asnis GM, Thomas M, Henderson MA. Pharmacotherapy treatment options for insomnia: a primer for clinicians. Int. J. Mol. Sci., 17, 50 (2015).
- 3) Ohayon MM, Roth T. Place of chronic insomnia in the course of depressive and anxiety disorders. J. Psychiatr. Res., 37, 9–15 (2003).
- 4) Breslau N, Roth T, Rosenthal L, Andreski P. Sleep disturbance and psychiatric disorders: a longitudinal epidemiological study of young adults. Biol. Psychiatry, 39, 411–418 (1996).
- 5) WHO. Depression. Fact sheet No. 369, October 2012.
- 6) Ford ES, Wheaton AG, Cunningham TJ, Giles WH, Chapman DP, Croft JB. Trends in outpatient visits for insomnia, sleep apnea, and prescriptions for sleep medications among U.S. adults: findings from the National Ambulatory Medical Care survey 1999–2010. Sleep, 37, 1283–1293 (2014).
- 7) Baba A. Barbiturates as hypnotics. Nihon Rinsho, 67, 1585–1589 (2009).
- 8) WHO. Chloral and chloral hydrate. IARC Monogr. Eval. Carcinog. Risks Hum., 63, 245–269 (1995).
- 9) Fukai Y, Ohta S. Hypnotic (barbiturate, non-barbiturate). Nihon Rinsho, 62 (Suppl. 12), 364–367 (2004).
- 10) Hajak G, Muller WE, Wittchen HU, Pittrow D, Kirch W. Abuse and dependence potential for the non-benzodiazepine hypnotics zolpidem and zopiclone: a review of case reports and epidemiological data. Addiction, 98, 1371–1378 (2003).
- 11) Interview form for Silece® tablets, eighth edition, revised March 2017 provided by Eisai Co., Ltd. (Tokyo, Japan).
- 12) Harada T, Fushimi K, Kato A, Ito Y, Nishijima S, Sugaya K, Yamada S. Demonstration of muscarinic and nicotinic receptor binding activities of distigmine to treat detrusor underactivity. Biol. Pharm. Bull., 33, 653–658 (2010).
- 13) Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br. Pharmacol. Chemother., 14, 48–58 (1959).
- 14) Interview form for Dalmate® capsules, third edition, revised March 2017 provided by Kyowa Pharmaceutical Industry Co., Ltd. (Osaka, Japan).
- 15) Interview form for Belsomra® tablets, sixth edition, revised November 2016 provided by MSD Co., Ltd. (Tokyo, Japan).
- 16) Patient information leaflet for Dalmane® capsules, revised May 2018 provided by Mylan Products Ltd. (Hertfordshire, U.K.).
- 17) Earl DE, Tietz EI. Inhibition of recombinant L-type voltage-gated calcium channels by positive allosteric modulators of GABAA receptors. J. Pharmacol. Exp. Ther., 337, 301–311 (2011).
- 18) Orzechowski RF, Currie DS, Valancius CA. Comparative anticholinergic activities of 10 histamine H1 receptor antagonists in two functional models. Eur. J. Pharmacol., 506, 257–264 (2005).
- 19) Blyden GT, Greenblatt DJ, Scavone JM, Shader RI. Pharmacokinetics of diphenhydramine and a demethylated metabolite following intravenous and oral administration. J. Clin. Pharmacol., 26, 529–533 (1986).
- 20) Pyle R, Scott M, Bartholomew J, McGrath S, Moffett B. Accidental polydipsia and hyponatremia from diphenhydramine urinary retention. Am. J. Med., 124, e5–e6 (2011).
- 21) Abraham O, Schleiden L, Albert SM. Over-the-counter medications containing diphenhydramine and doxylamine used by older adults to improve sleep. Int. J. Clin. Pharmacol., 39, 808–817 (2017).