
- |<
- <
- 1
- >
- >|
-
Takafumi Naito2019 年 42 巻 2 号 p. 149-157
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLTo date, limited drug information is available for the individual optimization of pharmacotherapy. The author attempted multiple evaluations of patient data on factors related to the pharmacokinetics, drug efficacy, and adverse reactions observed in clinical settings. Through the clinical studies, drug information on the individual optimization of pharmacotherapy needed by health professionals including physicians and pharmacists was identified. Major findings were: 1) Cachectic cancer patients had high plasma concentrations of oxycodone via the reduction of CYP3A activity. The metabolic reduction in cachectic cancer patients was potentially related to the elevated serum level of interleukin-6. 2) Dopamine receptor D2 (DRD2) genetic mutations and being female led to poor antiemetic efficacy of the treatment of opioid-induced nausea in prochlorperazine-treated patients. The opioid receptor μ1 (OPRM1) wild genotype in addition to being female and having high plasma concentrations of prochlorperazine increased prolactin secretion during oxycodone treatment. 3) Rheumatoid arthritis patients with a genetic mutation of ATP-binding cassette subfamily B member 1 (ABCB1) had high plasma concentrations of tacrolimus and its 13-O-demethylate. The ABCB1 genetic mutation and associated high plasma concentration of tacrolimus decreased kidney function. 4) Chronic inflammation increased the plasma voriconazole concentration via its poor metabolism, whereas it did not alter the plasma itraconazole concentration. Although co-administration of prednisolone did not affect the plasma concentration of triazole antifungals, it weakly increased voriconazole metabolism. 5) In breastfeeding women, the median milk/plasma concentration ratio of amlodipine was 0.85. However, the observed relative infant dose of amlodipine in most patients was less than 10%.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1022K) HTML形式で全画面表示
-
Junxian Ma, Xinbo Wang, Tingting Lv, Jie Liu, Ying Ren, Jinshan Zhang, ...2019 年 42 巻 2 号 p. 158-163
発行日: 2019/02/01
公開日: 2019/02/01
[早期公開] 公開日: 2018/11/15 ジャーナル フリー HTML
ジャーナル フリー HTMLGhrelin is a circulating peptide hormone, which involved in promoting feeding and regulating energy metabolism in human and rodents. Abnormal synovial hyperplasia is the most important pathologic hallmark of rheumatoid arthritis (RA), which is characterised by tumor-like expansion. Existing studies indicated that there may exist some relation between the decreased ghrelin and the abnormally proliferating synovial cells in RA. Therefore, the aim of this study is to explore the apoptotic effects of ghrelin on MH7A synovial cells in vitro. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to evaluate the effects of ghrelin on the viability of MH7A cells. Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick-end labeling (TUNEL) and flow cytometry were used to test the apoptotic effects of ghrelin. At last, Western blot and real-time PCR were performed to explore the expression of caspases-8, -9, and -3 after the treatment of ghrelin. MTT experiments showed that ghrelin could inhibit viability of MH7A cells. The results of flow cytometry and TUNEL showed that ghrelin could induce apoptosis of MH7A synovial cells. Western blot showed that expression of cleaved-caspases-8, -9, and -3 were increased in ghrelin stimulation group compared with the control group, while expression of pro-caspases-8, -9, and -3 had no significant difference. In mRNA levels, ghrelin can decrease pro-caspases-8, -9, and -3 mRNA expression, which confirmed the results of protein levels. Then these apoptotic effects were significantly reversed by [D-Lys3] GHRP-6 (ghrelin receptor antagonist). This study found that ghrelin can induce apoptosis of MH7A cells through caspase signaling pathways.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1904K) HTML形式で全画面表示 -
Hikari Suzuki, Kanan Bando, Hiroyuki Tada, Tomomi Kiyama, Takefumi Oiz ...2019 年 42 巻 2 号 p. 164-172
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLBisphosphonates (BPs) bind strongly to bone and exhibit long-acting anti-bone-resorptive effects. Among BPs, nitrogen-containing BPs (N-BPs) have far stronger anti-bone-resorptive effects than non-N-BPs. However, N-BPs induce acute inflammatory reactions (fever, arthralgia and myalgia, etc.) after their first injection. The mechanisms underlying these side effects remain unclear. Zoledronate (one of the most potent N-BPs) is given intravenously to patients, and the side-effect incidence is reportedly the highest among N-BPs. Our murine experiments have clarified that (a) intraperitoneally injected N-BPs induce various inflammatory reactions, including a production of interleukin-1 (IL-1) (a typical inflammatory cytokine), and these inflammatory reactions are weak in IL-1-deficient mice, (b) subcutaneously injected N-BPs induce inflammation/necrosis at the injection site, (c) lipopolysaccharide (LPS; a cell-wall component of Gram-negative bacteria) and N-BPs mutually augment their inflammatory/necrotic effects, (d) the non-N-BP clodronate can reduce N-BPs’ inflammatory/necrotic effects. However, there are few animal studies on the side effects of intravenously injected N-BPs. Here, we found in mice that (i) intravenous zoledronate exhibited weaker inflammatory effects than intraperitoneal zoledronate, (ii) in mice given intravenous zoledronate, LPS-induced production of IL-1α and IL-1β was augmented in various tissues, including bone, resulting in them increasing in serum, and (iii) clodronate (given together with zoledronate) prevented such augmentation and enhanced, slightly but significantly, zoledronate’s anti-bone-resorptive effect. These results suggest that infection may be a factor promoting the acute inflammatory side effects of N-BPs via augmented production of IL-1 in various tissues (including bone), and that clodronate may be useful to reduce or prevent such side effects.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (991K) HTML形式で全画面表示 -
Hiroyuki Takahashi, Sachio Okuda, Mizuho Tamura, Shintaro Kamei, Reiko ...2019 年 42 巻 2 号 p. 173-178
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLOptic neuritis is characterized by optic nerve inflammation, demyelination and axonal loss. Intravenous immunoglobulin (IVIg) has been reported to be effective for steroid-resistant patients. However, there is no report investigating the histopathological efficacy of IVIg in optic neuritis models. In this study, we examined the effects of IVIg on optic neuritis of experimental autoimmune encephalomyelitis (EAE) and experimental autoimmune optic neuritis (EAON). Inflammation, demyelination and axonal loss were assessed in the optic nerve sections. IVIg showed dose-dependent prevention of clinical symptoms in EAON. IVIg provided an anti-inflammatory effect in both EAE and EAON, associated with improved demyelination. Axonal loss in EAE was also significantly attenuated. These results suggest that IVIg has neuroprotective properties in experimental optic neuritis, and is a promising new treatment for optic neuritis.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (3877K) HTML形式で全画面表示 -
Songtao Wu, Naoki Uyama, Rei Atono Itou, Etsuro Hatano, Hiroko Tsutsui ...2019 年 42 巻 2 号 p. 179-186
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Daikenchuto (DKT) has been widely used for the treatment of postsurgical ileus in Japan. However, its effect on postsurgical adhesion formation has been obscure. In this study, the effect of DKT on postsurgical adhesion formation induced by cecum cauterization or cecum abrasion in mice was investigated. First, the expression of adhesion-related molecules in damaged ceca was investigated by quantitative (q)RT-PCR. During 24 h after surgery, mRNA expressions of interferon-γ (IFN-γ), plasminogen activator inhibitor-1 (PAI-1), interleukin-17 (IL-17), and Substance P (SP) in cauterized ceca and those of PAI-1 and IL-17 in abraded ceca were significantly up-regulated. Next, the effect of DKT on adhesion formation macroscopically evaluated with adhesion scoring standards. DKT (22.5–67.5 mg/d) was administered orally for 7 d during the perioperative period, and DKT did not reduce adhesion scores in either the cauterization model (control : DKT 67.5 mg/d, 4.8 ± 0.2 : 4.8 ± 0.2) or in the abrasion model (control : DKT 67.5 mg/d, 4.9 ± 0.1 : 4.5 ± 0.3). Histologically, collagen deposition and leukocyte accumulation were found at the adhesion areas of control mice in both models, and DKT supplementation did not alleviate them. Last, effect of DKT on expression of proadhesion moleculs was evaluated. DKT also failed to down-regulate mRNA expression levels of them in damaged ceca of both models. In conclusion, PAI-1 and IL-17 may be key molecules of postsurgical adhesion formation. Collagen deposition and leukocytes accumulation are histological characteristic feature of post-surgical adhesion formation. DKT may not have any preventive effect on postsurgical adhesion formation in mice.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (6660K) HTML形式で全画面表示 -
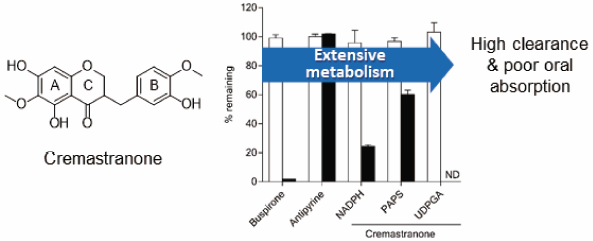
Eun-yeong Kim, Bit Lee, Seung-Yong Seo, Kiho Lee2019 年 42 巻 2 号 p. 187-193
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThe objective of this study was to characterize pharmacokinetics and metabolism of (±)-cremastranone (CMT) in mouse. Plasma concentrations of CMT following a single oral dose (10 mg/kg) were all below quantitation limit throughout 24-h time course, indicating poor oral bioavailability. Its plasma levels declined rapidly, with a half-life (t1/2) of 1.5 ± 0.3 min following a single intravenous dose (5 mg/kg). They were below the quantitation limit after 15 min post-dosing. CMT showed a high plasma clearance (CLp) of 7.73 ± 3.09 L/h/kg. Consistently, CMT was metabolized rapidly, with a t1/2 < 1 min when it was incubated with liver or intestine S9 fractions of mouse and human in the presence of cofactors for CYP450, uridine 5′-diphosphate (UDP)-glucuronosyltransferase (UGT), and sulfotransferase (ST). Further studies showed that CMT was metabolized by CYP450, UGT, and ST in vitro in liver S9 fractions of mouse and human, with UGT being the major enzyme responsible for its rapid metabolism. CMT was metabolized by UGT and ST in intestine S9 fractions of mouse and human. Mono-demethylated (M1), mono-glucuronide (M2), and mono-sulfate (M3 and M4) metabolites were tentatively identified in vitro. In conclusion, the pharmacokinetics of CMT is suboptimal as a systemic agent, especially as an oral therapy, due to its extensive metabolism. This report provides possible structural modifications to design CMT derivatives with better pharmacokinetic properties.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (897K) HTML形式で全画面表示 -
Li-Hua Mu, Hong Yan, Yu-Ning Wang, Teng-Fei Yu, Ping Liu2019 年 42 巻 2 号 p. 194-200
発行日: 2019/02/01
公開日: 2019/02/01
[早期公開] 公開日: 2018/11/21 ジャーナル フリー HTML
ジャーナル フリー HTMLSeventeen 13,28-epoxy triterpenoid saponins obtained from Ardisia gigantifolia STAPF. were evaluated their anti-proliferative activities on MCF-7 cells. The structure–activity relationship analysis indicated that CH3 group at C-30, four saccharide units with L-rhamnose at R6 in the sugar units are crucial for the cytotoxic activity on MCF-7. Compounds 1, 2, 6, 7, 12, and 14 were selected to identify the anti-proliferative activity on the other three breast cancer cell lines (T47D, MDA-MB-231 and SK-BR-3). Compounds 2, 6, and 7 with good activity on MCF-7 also showed activity on T47D, MDA-MB-231, and SK-BR-3. Compounds 12 and 14 without cytotoxic activity on MCF-7 almost showed no activities on the other three cell lines. For the triple-negative breast cancer MDA-MB-231, Saponins 7 and 14 showed selective cytotoxic activity, 7 showed much more activity than 14, suggesting the six saccharide units in sugar units and CH3 on C-30 were the key moieties for the anti-proliferative activities. Further molecular mechanism of saponin 7 was studied on inhibiting cell proliferation of MDA-MB-231 cells. Saponin 7 could enhance apoptosis, arrest cell cycles, decrease mitochondrial membrane potentials (MMPs), and considered the involvement of reactive oxygen species (ROS) may explain this conundrum.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1397K) HTML形式で全画面表示 -
Elian Alegría-Herrera, Maribel Herrera-Ruiz, Rubén Román-Ramos, Alejan ...2019 年 42 巻 2 号 p. 201-211
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThe main objective of treatment against hypertension is not only to reduce blood pressure levels, but also to reduce vascular risk in general. In the present work, administering angiotensin II (AGII; 0.2 µg/kg intraperitoneally (i.p.) for 12 weeks) activates the hypothalamic–pituitary–adrenal (HPA) axis, which caused an increase in corticosterone levels, as well as in proinflammatory cytokines (interleukin 1β (IL-1β), interleukin 6 (IL-6), and tumor necrosis factor alpha (TNF-α)) and macrophage chemotactic protein 1 (MCP-1), and decreased anti-inflammatory cytokines (interleukin 10 (IL-10) and interleukin 4 (IL-4)). On observing the behavior in the different models, an anxiogenic effect (elevated plus maze (EPM)) and cognitive impairment (water Morris maze (WMM)) was observed in animals with AGII. By administering organic extracts from Ocimum basilicum (Oba-EtOAc) and Ocimum selloi (Ose-EtOAc), and some doses of rosmarinic acid (RA) (6 weeks per os (p.o.)), the damage caused by AGII was stopped by re-establishing corticosterone serum levels and by decreasing the proinflammatory cytokines and MCP-1.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2044K) HTML形式で全画面表示 -
Akiko Takagaki, Yasukiyo Yoshioka, Yoko Yamashita, Tomoya Nagano, Masa ...2019 年 42 巻 2 号 p. 212-221
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLGlucose uptake ability into L6 skeletal muscle cell was examined with eleven kinds of ring fission metabolites of (−)-epigallocatechin gallate (EGCG) produced by intestinal bacteria. The metabolites 5-(3,5-dihydroxyphenyl)-γ-valerolactone (EGC-M5), 4-hydroxy-5-(3,4,5-trihydroxyphenyl)valeric acid (EGC-M6), 5-(3,4,5-trihydroxyphenyl)-γ-valerolactone (EGC-M7) and 5-(3-hydroxyphenyl)valeric acid (EGC-M11) have been found to promote uptake of glucose into L6 myotubes significantly. EGC-M5, which is one of the major ring fission metabolites of EGCG, was also found to have a promotive effect on glucose transporter 4 (GLUT4) translocation accompanied by phosphorylation of AMP-activated protein kinase (AMPK) signaling pathway in skeletal muscle both in vivo and in vitro. Furthermore, the effect of oral single dosage of EGC-M5 on glucose tolerance test with ICR mice was examined and significant suppression of hyperglycemia was observed. These data suggested that EGC-M5 has an antidiabetic effect in vivo.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1165K) HTML形式で全画面表示 -
Shenghui Huang, Ting Gong, Tengfei Zhang, Xinfeng Wang, Qianqian Cheng ...2019 年 42 巻 2 号 p. 222-230
発行日: 2019/02/01
公開日: 2019/02/01
[早期公開] 公開日: 2018/12/05 ジャーナル フリー HTML
ジャーナル フリー HTMLZhongfenggao (ZFG) is prescribed for the treatment of cerebrovascular diseases in critical projects of the State Administration of Traditional Chinese Medicine. ZFG has been found to nourish qi, activate blood circulation, remove blood stasis, dredge collaterals, and strengthen the brain and mind. The present study investigated the effects of ZFG on oxygen–glucose deprivation–reoxygenation (OGD/R) induced injury to brain microvascular endothelial cells (BMECs), and the mechanisms underlying such effects. BMECs are essential target cells of ischemic stroke. In order to simulate ischemic-like conditions in vitro, BMECs were exposed to glucose deprivation and hypoxia for 2 h. Results indicate that ZFG may protect OGD/R-induced injury to BMECs by promoting angiogenesis. Further, we observed that ZFG significantly inhibited apoptosis induced by OGD/R injury. ZFG significantly promoted migration and microtubule formation in BMECs under OGD/R conditions. Additionally, ZFG increased levels of the vascular endothelial growth factor (VEGF) significantly and activated the Notch and Wnt signaling pathways. The results of the present study indicate that ZFG may display a protective effect against OGD/R-induced BMECs injury by promoting angiogenesis via Notch and Wnt signaling pathways. These results provide novel insights into the mechanisms underlying the therapeutic action of ZFG which shows promise as a potential drug candidate for treating cerebral ischemia–reperfusion.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (3037K) HTML形式で全画面表示 -
Jiangang Chen, Wenfang Lu, Hao Chen, Xiaoli Bian, Guangde Yang2019 年 42 巻 2 号 p. 231-246
発行日: 2019/02/01
公開日: 2019/02/01
[早期公開] 公開日: 2018/11/30 ジャーナル フリー HTML
ジャーナル フリー HTMLIn this study, a series of salicylic acid derivatives were designed and synthesized as novel non-saccharide α-glucosidase inhibitors. Biological evaluation indicated that when compared to acarbose, compounds T9, T10, and T32 exhibited a higher potency of α-glucosidase inhibitory activity with IC50 values of 0.15 ± 0.01, 0.086 ± 0.01 and 0.32 ± 0.02 mM, respectively. Evaluation of the inhibition kinetics indicated that T9, T10, T32, and acarbose interacted with α-glucosidase in a mixed non-competitive inhibitory manner. Moreover, T9, T10, and T32 statically quenched the fluorescence of α-glucosidase by formation of an inhibitor-α-glucosidase complex. The docking results showed that hydrogen bonds were generated between the test compounds and α-glucosidase. The antioxidant study revealed that compound T10 exhibited a higher antioxidant activity via scavenging 1,1-diphenyl-2-picrylhydrazyl free radical (DPPH), thereby inhibiting lipid peroxidation and the total reduction capacity. In brief, the salicylic acid derivatives identified in this study were promising candidates for development as novel non-saccharide α-glucosidase inhibitors.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2020K) HTML形式で全画面表示 -
Risa Akizuki, Hiroaki Eguchi, Satoshi Endo, Toshiyuki Matsunaga, Akira ...2019 年 42 巻 2 号 p. 247-254
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLAbnormal expression of the tight junctional components claudins (CLDNs) is observed in various malignant tissues. We reported recently that CLDN18 expression is down-regulated in human lung adenocarcinoma tissues. In the present study, we investigated the biological functions of CLDN18 using lung adenocarcinoma A549 cells. Microarray analysis showed that CLDN18 increases zonula occludens (ZO)-2 expression in A549 cells. The ectopic expression of CLDN18 increased nuclear ZO-2 levels, which were inhibited by N-[2-[[3-(4-bromophenyl)-2-propen-1-yl]amino]ethyl]5-isoquinolinesulfonamide (H-89), a nonspecific protein kinase A (PKA) inhibitor, but not by a PKA inhibitor 14-22 amide. In addition, dibutyryl cyclic adenosine monophosphate, an analogue of PKA, did not increase ZO-2 levels. These results suggest that H-89 sensitive factors without PKA are involved in the CLDN18-induced elevation of ZO-2. The cell cycle was affected by neither ZO-2 knockdown in CLDN18-expresssing A549 (CLDN18/A549) cells nor ZO-2 overexpression in A549 cells, suggesting that ZO-2 does not play an important role in the regulation of cell proliferation. The introduction of ZO-2 small interfering RNA (siRNA) into CLDN18/A549 cells increased migration, the expression and activity of matrix metalloproteinase 2 (MMP2), and the reporter activity of an MMP2 promoter construct. Furthermore, H-89 enhanced both mRNA levels and reporter activity of MMP2 in CLDN18/A549 cells. These results suggested that a reduction in CLDN18-dependent ZO-2 expression enhances MMP2 expression in lung adenocarcinoma cells, resulting in the promotion of the cell migration. CLDN18 may be a novel marker for metastasis in lung adenocarcinoma.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (3015K) HTML形式で全画面表示 -
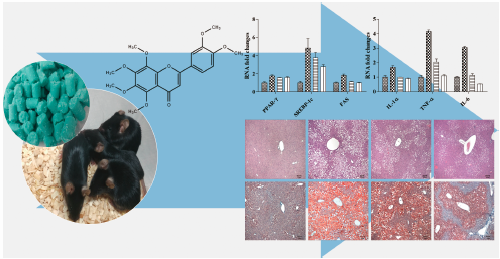
Hyoung-Yun Han, Sung-Kwon Lee, Bong-Keun Choi, Dong-Ryung Lee, Hae Jin ...2019 年 42 巻 2 号 p. 255-260
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLNonalcoholic fatty liver disease (NAFLD) is characterized by hepatic lipid accumulation, which is the most common form of chronic liver disease. Multiple clinical studies using natural compounds such as flavonoids have been conducted to treat NAFLD. In the present study, the pharmacological effect of Citrus aurantium L. (Rutaceae) peel extract (CAE), which contains over 27% of polymethoxyflavone nobiletin, on NAFLD was evaluated using a high-fat diet (HFD) animal model susceptible to developing NAFLD. C57BL/6 mice were fed an HFD (60% kcal of energy derived from fat) for 8 weeks to induce obesity. Obese mice were randomly allocated to four groups of eight mice each (HFD alone, HFD with silymarin, HFD with 50 mg/kg CAE, and HFD with 100 mg/kg CAE). After 8 weeks of treatment, all mice were euthanized, and plasma and liver tissues were analyzed biochemically and histopathologically. The results indicate that CAE treatment significantly reduced HFD-induced NAFLD, as shown by decreased serum lipid index and prevented liver histopathology. The expression of genes involved in lipid synthesis including free fatty acid (FFA), peroxisome-proliferator-activated receptor γ (PPAR-γ), sterol receptor element binding protein 1c (SREBP-1c), and fatty acid synthesis enzyme was suppressed by CAE treatment. Moreover, compared to untreated mice, CAE-treated HFD mice showed decreased pro-inflammatory cytokine expression. These results demonstrated that CAE prevented HFD-induced NAFLD by reducing plasma levels of triglyceride and cholesterol and de novo lipid synthesis.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1420K) HTML形式で全画面表示 -
Peng Li, Shijie Wang, Huiqin Wang, Hong Yan2019 年 42 巻 2 号 p. 261-267
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLA series of tetraethyl 2,4,8,10-tetramethyl-6,12-diaryl-3,9-dioxahexacyclo[6.4.0.02,7.04,11.05,10]dodecane-1,5,7,11-tetracarboxylates (simplified as 3,9-dioxatetraasteranes) with C2-symmetric structural characteristics were synthesized through the [2 + 2] photocycloaddition of the diethyl 2,6-dimethyl-4-aryl-4H-pyran-3,5-dicarboxylates. Besides, their anti-human immunodeficiency virus (HIV)-1 activities were evaluated by enzyme-linked immunosorbent assay (ELISA) assay against HIV-1 (IIIB) replication in MT-4 cell culture. The result showed that the tested compounds exhibited potential activates with IC50 values less than 110 nM. Furthermore, docking study was carried out to study the binding mode of these compounds. The results indicated that the overall orientation of the inhibitors in the active site were similar to that of the cyclic urea AHA001 and a hydrogen bond with the protein residues might play a crucial role in their anti-HIV-1 activities. Such results will provide a theoretical foundation for further investigations on the biological activity of 3,9-dioxatetraasteranes.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1543K) HTML形式で全画面表示 -
Naoki Agata, Yoshimitsu Kato, Shogo Hamaguchi, Iyuki Namekata, Hikaru ...2019 年 42 巻 2 号 p. 268-272
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThe presence and function of the ATP-sensitive potassium channel current (IKATP) were examined in the guinea pig myocardium to clarify the mechanisms for the resistance of the fetal myocardium to hypoxia. Experimental hypoxia markedly reduced the action potential duration and contractile force in isolated ventricular myocardium from the adult, but only moderately in those from the fetus. In isolated ventricular cardiomyocytes, the density of the IKATP activated by cromakalim, as well as their sensitivity to intracellular ATP concentration, were not different between the fetus and adult. The tissue ATP content was similar between the fetal and adult myocardium under normal condition, but the hypoxia-induced decrease was smaller in the fetus. Confocal microscopic analysis revealed that the mitochondria in the fetal cardiomyocyte is less in quantity than that in the adult and is more localized to the cell center. These results indicate that IKATP in the fetal guinea pig myocardium has a current density and ATP sensitivity similar to those of the adult, but is not activated under hypoxic conditions because the energy metabolism of the fetal myocardium is less dependent on oxidative phosphorylation.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (827K) HTML形式で全画面表示 -
 Kosuke Sakai, Hidemasa Katsumi, Kentaro Kamano, Kiyo Yamauchi, Ayuko H ...2019 年 42 巻 2 号 p. 273-279
Kosuke Sakai, Hidemasa Katsumi, Kentaro Kamano, Kiyo Yamauchi, Ayuko H ...2019 年 42 巻 2 号 p. 273-279
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Hydrogen sulfide (H2S) is an endogenous gaseous transmitter known to play an important role in biological functions. For the hepatic and intrahepatic targeting of H2S prodrug at the cellular level, we developed two types of sulfo-albumins, in which five sulfide groups (source of H2S) were covalently bound to succinylated (Suc) or galactosylated (Gal) bovine serum albumin (BSA). Sulfo-BSA-Suc and polyethylene glycol (PEG)-Sulfo-BSA-Gal, both released H2S in the 5 mM glutathione solution, but not in the plasma. Sulfo-BSA-Suc and PEG-Sulfo-BSA-Gal were taken up by RAW264.7 cells (mouse macrophage-like cells) and Hep G2 cells (human hepatocellular carcinoma cells), respectively, and H2S was released. These results indicate that Sulfo-BSA-Suc and PEG -Sulfo-BSA-Gal selectively released H2S intracellularly. In a biodistribution study, up to 80% of 111In-labeled Sulfo-BSA-Suc and PEG-Sulfo-BSA-Gal rapidly accumulated in the liver, 30 min after intravenous injection in mice. Furthermore, 111In-labeled Sulfo-BSA-Suc and PEG-Sulfo-BSA-Gal predominantly accumulated in liver nonparenchymal (endothelial cells and Kupffer cells) and parenchymal cells (hepatocytes), respectively. These findings suggest that targeted delivery of H2S prodrug to a specific type of liver cells was successfully achieved by bioconjugation.
Graphical Abstract Fullsize Image抄録全体を表示Editor's pickThe delivery of hydrogen sulfide (H2S) to liver is expected for the treatment of hepatic diseases. K. Sakai et al. developed two types of sulfo-albumins, macromolecular H2S prodrugs, for hepatic and intrahepatic targeting of H2S. Sulfide groups (source of H2S) were covalently bound to succinylated (Suc) and galactosylated (Gal) bovine serum albumin (BSA) for targeted delivery of H2S to hepatic nonparenchymal cells and parenchymal cells, respectively. Their results demonstrated targeted delivery of H2S prodrug to a specific type of liver cells using the chemical modification of targeting ligands.
PDF形式でダウンロード (2767K) HTML形式で全画面表示 -
Keisuke Obara, Lin Ao, Tsukasa Ogawa, Takumi Ikarashi, Fumiko Yamaki, ...2019 年 42 巻 2 号 p. 280-288
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThe present study aimed to investigate the potential inhibitory effects of 21 clinically available hypnotics on acetylcholine (ACh)-induced contractions in rat urinary bladder smooth muscle (UBSM) in order to predict whether these hypnotics could induce voiding impairment. ACh-induced contraction in rat UBSM was inhibited only by diphenhydramine (a histamine H1 receptor antagonist) at a concentration that was clinically relevant. ACh-induced contraction was also significantly inhibited by flurazepam (a benzodiazepine hypnotic) and suvorexant (an orexin receptor antagonist), albeit at concentrations that substantially exceeded clinically achievable blood levels. These three drugs (at 10−5 M) also inhibited high-KCl (80 mM) Locke–Ringer solution-induced contractions. In contrast to the effects of the abovementioned hypnotics, ACh-induced contractions were not significantly affected by triazolam, etizolam, brotizolam, lormetazepam, estazolam, flunitrazepam, nitrazepam (benzodiazepine hypnotics), thiopental, thiamylal, pentobarbital, amobarbital, secobarbital, phenobarbital (barbiturate hypnotics), zolpidem (an imidazopyridine hypnotic), zopiclone (a cyclopyrrolone hypnotic), ramelteon (a melatonin receptor agonist), bromovalerylurea, and chloral hydrate. These findings suggest that most clinically used hypnotics are not likely to result in anticholinergic-induced dysuria within their clinically achievable blood concentration ranges. Diphenhydramine may, however, induce voiding impairment, an action attributable to diminished UBSM contractility within its clinical dose range.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1263K) HTML形式で全画面表示
-
Tomohiro Fuse, Akio Yanagida, Mitsuhiro Shimizu2019 年 42 巻 2 号 p. 289-294
発行日: 2019/02/01
公開日: 2019/02/01
[早期公開] 公開日: 2018/12/07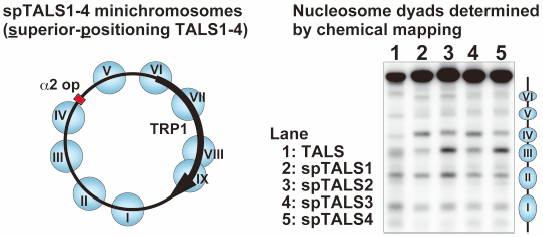 ジャーナル フリー HTML
ジャーナル フリー HTMLIn eukaryotic genomes, the nucleosome is the structural and functional unit, and its position and dynamics are important for gene expression control and epigenetic regulation. Epigenetics is an important mechanism in development and homeostasis, and aberrant epigenetics is a common feature in cancer. Although understanding the mechanistic basis that determines nucleosome positioning in vivo is important for elucidating chromatin function and epigenetic regulation, a suitable experimental system to examine such mechanisms is still being developed. Herein, we examined nucleosome organization in yeast minichromosomes, using a parallel mapping method we previously developed that involve site-directed chemical cleavage and micrococcal nuclease digestion. This parallel mapping is capable of revealing the differences in the occupancy and the stability of individual nucleosomes in the minichromosome. Based on the previously characterized minichromosome, we engineered a set of new minichromosomes, aimed at strengthening the positioning of the nucleosomes. The site-directed chemical mapping method demonstrated that the nucleosome positioning in the newly designed yeast minichromosome system was significantly more stable. This system will be useful for elucidating the determinants of nucleosome organization, such as DNA sequences and/or nucleosome binding proteins, and for determining the relationships between nucleosome dynamics and epigenetic regulation, which are targets for therapeutic agents.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1326K) HTML形式で全画面表示 -
Makiko Fujii, Kiku Kato, Miko Imai, Hiroki Kuwabara, Minori Awano, Kan ...2019 年 42 巻 2 号 p. 295-298
発行日: 2019/02/01
公開日: 2019/02/01
[早期公開] 公開日: 2018/12/01 ジャーナル フリー HTML
ジャーナル フリー HTMLSurface free energy (SFE) is an important factor for evaluation of wettability or adhesion. Thus, the SFE of a Yucatan micropig (YMP) skin and a hairless mouse (HM) skin, which are commonly used in skin permeation studies instead of human skin, were compared with the human skin. Contact angles of water and 1-bromo naphthalene to skin were measured and the SFE was calculated using the Owens–Wendt equation. The SFE of the human abdominal skin was 40 mN/m and its polar component σsp was as low as 2 mN/m, which was similar to that of the low sebum skin reported previously. In the case of the YMP skin, σsp was high on the surface but similar to that obtained after the skin was tape-stripped twice. The HM skin showed similar SFE as that of the human skin. When the surfactant was applied on the skin, wiped, and dried, the remaining surfactant increased the SFE in σsp; however, the original SFE was obtained after rinsing with water. The YMP skin and HM skin is similar to the human abdominal skin with a low sebum level. Thus, they are also good skin models for studying wettability or adhesion of a substance.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (476K) HTML形式で全画面表示 -
Takara Ohto, Manami Konishi, Hiroki Tanaka, Koji Onomoto, Mitsutoshi Y ...2019 年 42 巻 2 号 p. 299-302
発行日: 2019/02/01
公開日: 2019/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録While the use of in vitro-transcribed mRNA (IVT-mRNA) in therapeutics is a rapidly expanding area, the transfection of the exogenous IVT-mRNA is accompanied by a risk of immune activation. This immunological defense mechanism suppresses cellular translation process and can reduce transfection efficiency to a considerable extent. In the present study, we investigated the in vitro effects of Integrated Stress Response Inhibitor (ISRIB), and dexamethasone, a steroidal anti-inflammatory drug, on the transfection activity of a lipid nanoparticle (LNP) that was composed of ionizable lipids and IVT-mRNA. In the case of transfection to mouse embryonic fibroblast (MEF) cells, ISRIB mainly enhanced the transfection activity at an early stage of transfection (0–6 h). In contrast, dexamethasone caused an increase in transfection activity at intermediate-late stages of transfection (4–48 h). We also investigated the in vivo effects of dexamethasone using an LNP on that the IVT-mRNA and lipid-conjugated dexamethasone (Dex-Pal) were co-loaded. The intravenous administration of the LNP successfully enhanced the protein expression in a mouse liver by up to 6.6-fold. Collectively, the co-delivery of an anti-inflammatory drug is a promising approach for enhancing transfection efficiency of IVT-mRNA.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (431K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|