Regular Articles
Expression of Active Staphylococcus aureus Tyrosine Kinases in a Human Cell Line
2019 年 42 巻 3 号 p. 411-416
詳細
2019 年 42 巻 3 号 p. 411-416
Many bacteria encode tyrosine kinases that are structurally unrelated to their eukaryotic counterparts and are termed BY-kinases. Two BY-kinases, CapB1 and CapB2, have been identified in the Staphylococcus aureus genome. Although CapB1 and CapB2 share more than 70% homology, earlier studies with purified enzymes did not find any evident kinase activity in CapB1, whereas CapB2 was autophosphorylated on a C-terminal tyrosine cluster in the presence of the kinase modulator proteins CapA1 or CapA2. For the convenient analysis of BY-kinases, we attempted to express CapB2 in an active form in a mammalian cell line. To this end, the C-terminal activation domain of CapA1 was attached to the N-terminus of CapB2, and the resulting CapA1/CT-CapB2 chimera was further fused with various tags and transfected into HEK293T cells. Immunoblotting analyses showed that when fluorescent protein tags were attached to the N-terminus, CapA1/CT-CapB2 was both expressed and tyrosine phosphorylated in HEK293T cells. Mutation of the ATP-binding lysine abrogated tyrosine phosphorylation, indicating that tyrosine phosphorylation was catalyzed by the transfected bacterial kinase and not by endogenous cellular enzymes. Unexpectedly, mutation of the C-terminal tyrosine cluster did not abolish autophosphorylation. Further analyses revealed that CapA1/CT-CapB2 phosphorylated not only itself but also the attached fluorescent protein tag. Several domains and residues important for tyrosine kinase activity were identified from the production of various mutants. We also present data that CapB1, which was previously thought to be catalytically inert, may possess intrinsic kinase activity.
A variety of bacteria possess genes that encode tyrosine kinases named BY-kinases, which share no structural similarities with their eukaryotic counterparts.1–4) BY-kinases do not contain the 11 Hanks motif of eukaryotic kinases but are characterized by three Walker-type motifs found in many ATP/guanosine 5′-triphosphate (GTP)-binding proteins and a C-terminal tyrosine cluster that is the site of autophosphorylation.
BY-kinases are divided into two subfamilies. Gram-positive bacterial BY-kinases are composed of two proteins, a membrane-bound activator and a cytoplasmic catalytic module. The activator contains two membrane-spanning domains and a short cytoplasmic C-terminal tail that interacts with—and activates—the cytoplasmic kinase. In Gram-negative bacteria, the activation and the catalytic domains are fused and expressed as a single polypeptide.
BY-kinases have been implicated in various events, including stress responses, the cell cycle, and DNA replication,5–8) but their best-studied function is their essential contribution to polysaccharide synthesis and transport. The majority of BY-kinases are encoded in the operons that regulate polysaccharide production. In Staphylococcus aureus, the BY-kinase CapB2 phosphorylates and activates the uridine 5′-diphosphate (UDP)-N-acetyl-mannosamine dehydrogenase Cap5O, a key enzyme in capsule formation.9) Likewise, Escherichia coli and Bacillus subtilis BY-kinases phosphorylates UDP-dehydrogenases required for exopolysaccharide synthesis.10,11) The involvement of BY-kinases in polysaccharide production has been reported in various other bacteria as well.12–14)
Bacterial polysaccharides are considered important virulence factors, and inhibition of BY-kinase function is thus expected to affect bacterial growth and pathogenicity. Since BY-kinases do not share any similarities with eukaryotic counterparts, they are considered attractive drug targets for novel antibiotics.15–17) However, their characterization and inhibitor screening have both been hampered by the lack of versatile methods to monitor kinase activity. The assays for BY-kinases described to date rely on in vitro reactions using purified enzymes and radioactive 32P-ATP. The number of kinases actually tested for activity has also been limited, and the majority of the sequences annotated as BY-kinases are based on the existence of signature motifs. Thus, the assignments remain putative.
To reduce the time and effort required for protein purification and radioisotopic assays, we attempted to express BY-kinases in mammalian cell lines and detect their activity by immunoblotting. One BY-kinase that has been assayed to date is S. aureus CapB2. The S. aureus genome codes for two BY- kinases, CapB1 and CapB2, as well as their cognate activators, CapA1 and CapA2.18) Although the kinase activity of CapB1 has not been demonstrated, CapB2 exhibits tyrosine kinase activity in vitro. The kinase activity of CapB2 was also significantly enhanced when it was fused to the C-terminal domain of CapA protein.18–20)
Here, we show that the S. aureus BY-kinase CapB2, and possibly CapB1, can be translated into active tyrosine kinases in a human cell line when they are fused to CapA C-terminal cytoplasmic activation domains. This approach provides an efficient and economical method to measure and analyze BY-kinases, and could be useful in future enzyme characterization and inhibitor screening.
Rabbit anti-CapB2 antibody was raised against amino acids 183–199 of CapB2. Anti-phosphotyrosine antibodies 4G10 and PY20 were from Merck Millipore (U.S.A.) and Santa Cruz (U.S.A.), respectively. Anti-red fluorescent protein (RFP) monoclonal and polyclonal antibodies and anti-RFP magnetic beads were products of Medical & Biological Laboratories (Nagoya, Japan). TransIT293 was from Mirus Bio (U.S.A.), and PreScission HRV3C protease was from GE Healthcare (U.S.A.).
PlasmidsThe Gateway pDONR221 donor vector and destination vectors for expression of emerald green fluorescent protein (EGFP)-, glutathione S-transferase (GST)-, 6xHistidine (6xHis)-, V5- and yellow fluorescent protein (YFP)-tagged proteins in mammalian cells were products of Invitrogen (U.S.A.). Other destination vectors—including those with DsRed-Express (DsRedEx) and other fluorescent protein tags, or the N-terminal 10 amino acids of mouse Lck (LckN) for membrane localization21)—were created in our laboratory by modification of the purchased vectors.
CapA1, CapA2, CapB1, and CapB2 DNAs were isolated by PCR from genomic DNA of S. aureus ATC C6538P. CapA/CT-CapB chimeras were constructed by the fusion of DNA fragments coding for the C-terminal 29 and 28 amino acids of CapA1 and CapA2, respectively, with full-length CapB1 or CapB2 DNA. They were then cloned into a pDONR221 vector to create Gateway entry clones. Mutations were introduced in entry clones using standard recombinant DNA technology. The CapA/CT-CapB chimeric genes were moved to various destination vectors to generate mammalian expression plasmids according to the manufacturer’s instructions.
Immunoblotting Analysis of S. aureus Tyrosine Kinases Expressed in HEK293T CellsHEK293T cells were suspended in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and seeded in collagen-coated 96-well plates at a density of 2.5 × 104 cells per well. After attachment, cells were transfected with 100 ng expression plasmids using TransIT293 reagent. Two days post-transfection, the cells were fixed with 10% trichloroacetic acid and then lysed with 9 M urea, 2% Triton-X-100, and 2% lithium dodecyl sulfate. Lysates were neutralized with 2 M Tris and subjected to immunoblotting.
To analyze phosphorylation domains, HEK293T cells were transfected with expression plasmids into which the eight-amino acid human rotavirus 3C (HRV3C) protease recognition sequence was inserted between the N-terminal DsRedEx-tag and the CapA1/CT-CapB2 chimeras. The transfected cells were cultured for two days and then lysed with 20 mM Tris–HCl, pH 7.5 containing 0.5% Triton-X 100, 0.5% 3-[(3-Cholamidopropyl)dimethylammonio]propanesulfonate (CHAPS), 150 mM NaCl, 0.1 mM Na3VO4, and protease inhibitors. The lysates were centrifuged, and anti-RFP magnetic beads were added to the supernatants. After incubation at 4°C for three hours, the beads were washed and suspended in 20 mM Tris–HCl, pH 7.5, 150 mM NaCl, and 1 mM dithiothreitol. The beads were treated with HRV3C protease at 4°C for 16 h to cleave off the CapA1/CT-CapB2 segments, and the DsRedEx-tag remaining bound on the beads was then eluted by the addition of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer.
BY-kinases have been assayed by classical methods using purified proteins and radioactive ATP.11,18,22) To develop more convenient BY-kinase assays, we tested whether bacterial kinases can be expressed in mammalian cell lines and detected their activity by immunoblotting. Previous reports have shown that the activity of the S. aureus BY-kinase CapB2 is enhanced when it is fused to the C-terminal cytoplasmic tail of the activator CapA1.18–20) Based on these reports, we constructed CapA1/CT-CapB2 by attaching the C-terminal 29 amino acids of CapA1 to the N-terminus of full-length CapB2 (Fig. 1A). CapA1/CT-CapB2 was then moved to different expression vectors that we either purchased or generated in our laboratory. The expression clones were transfected into HEK293T cells and the production of CapA1/CT-CapB2 protein monitored by immunoblotting using anti-CapB2 antibody (Fig. 2, CapB2).

LckN, N-terminal 10 amino acids of mouse Lck; HRV3C, human rotavirus 3C protease recognition sequence, A. Positions of the CapA C-terminal cytoplasmic tail (CapA/CT), EX2RX2R, Walker A (A), Walker A′ (A′), Walker B (B), lysine 55 (K55), valine 127 (V127), and the C-terminal tyrosine cluster (CTY) are shown.

Expression plasmids were transfected into HEK293T cells and subjected to immunoblotting using anti-CapB2 (CapB2) and anti-phosphotyrosine (PY) antibodies. p62/PB1, p62/SQSTM1 PB1 domain; GST, Glutathione S-transferase; MAT, metal affinity tag; LckN, N-terminal 10 amino acids of mouse Lck; BCR, breakpoint cluster region protein; NPM, nucleophosmin.
The expression of CapA1/CT-CapB2 protein in HEK293T cells was significantly influenced by the tags attached. Commonly used tags, such as GST, 6xHis, and V5, or the oligomerizing protein tags we produced (p62/PB1, BCR/1-72, ETV6/1-155, and NPM/1-117) did not support expression of CapA1/CT-CapB2 when fused to the N-terminus.
In contrast, CapB2 was expressed in nine out of the ten N-terminal fluorescent protein tags tested (AcGFP, Azami Green, DsRed-Monomer, DsRedEx, mCherry, EGFP, Keima Red, Monti-Red, tdTomato, and YFP), with Keima Red being the exception. We also attached EGFP, YFP, and Azami Green to the C-terminus of CapA1/CT-CapB2, but only the Azami Green-tagged protein was detected. The complementary split-fluorescent protein fragments Kaede Green-N and Kaede Green-C were ineffective for CapA1/CT-CapB2 expression.
The samples were then analyzed for tyrosine phosphorylation (Fig. 2, PY). Many of the N-terminal fluorescent protein-tagged CapA1/CT-CapB2 reacted with anti-phosphotyrosine antibodies, raising the possibility that CapA1/CT-CapB2 can be translated into an active tyrosine kinase in HEK293T cells. For further studies, we selected the LckN-DsRedEx-tag, in which the N-terminal dual acylation motif of mouse Lck directs the fused protein to the membrane and the DsRedEx facilitates visualization by fluorescence microscopy.
To confirm that CapA1/CT-CapB2 was autophosphorylated and not the substrate of host cell kinases, the ATP-binding lysine in the Walker A domain (lysine 55) was mutated to methionine (CapA1/CT-CapB2/K55M), and the four C-terminal tyrosines reported to be the site of autophosphorylation (tyrosines 221, 222, 224, 225) were replaced with phenylalanines (CapA1/CT-CapB2/4F). Tyrosine phosphorylation was completely abrogated in the K55M mutant (Fig. 3, lane 2), suggesting that phosphorylation was due to the activity of CapA1/CT-CapB2 itself, and not of endogenous cellular kinases.
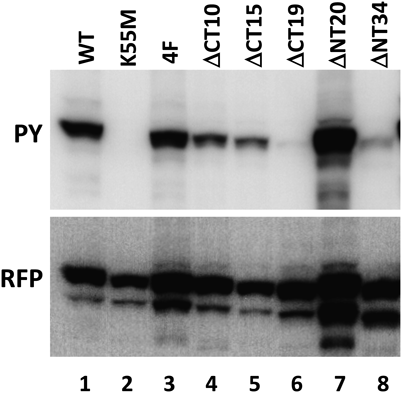
HEK293T cells were transfected with plasmids to express wild-type (WT) and mutant CapA1/CT-CapB2 proteins fused to LckN-DsRedEx and subjected to immunoblotting using anti-phosphotyrosine (PY) and anti-RFP (RFP) antibodies.
Somewhat unexpectedly, and in contradiction to a previous report,19) mutation of the four C-terminal tyrosines to phenylalanines resulted in no reduction of the phosphotyrosine signal (Fig. 3, lane 3). We were not completely confident about this result, and so we made C-terminal deletions of 10, 15, and 19 amino acids to remove the entire tyrosine cluster region (Fig. 3, lanes 4, 5, 6). Although phosphorylation decreased significantly after the removal of 19 residues, the 15-amino-acid deletion mutant was still phosphorylated. To explore the possibility of phosphorylation of other tyrosine residues in CapB2, we mutated each upstream tyrosine, but none of the mutants exhibited decreased reactivity with anti-phosphotyrosine antibodies (data not shown).
The most plausible explanation for the above results seemed to be tyrosine phosphorylation of the DsRedEx-tag by CapA1/CT-CapB2/4F, although, to our knowledge, fluorescent proteins have never been reported to be targets of tyrosine phosphorylation. To assess the phosphorylation of DsRedEx, we introduced the eight-amino-acid human rotavirus 3C (HRV3C) protease site (LEVLFQGP) to the C-terminus of DsRedEx so that the BY-kinase and the fluorescent protein tag moieties could be separated by protease treatment (Fig. 1B).
Wild-type CapA1/CT-CapB2 and the CapA1/CT-CapB2/4F mutant with the cleavable DsRedEx-tag were expressed in HEK293T cells and collected on anti-RFP-coated magnetic beads. The beads were then treated with HRV3C protease to release the CapA1/CT-CapB2 domain, leaving the DsRedEx on the beads for elution by the addition of SDS-PAGE sample buffer. Both fractions were subjected to immunoblotting using anti-phosphotyrosine, anti-CapB2, and anti-RFP antibodies.
As shown in Fig. 4, DsRedEx-tagged wild-type CapA1/CT-CapB2 was phosphorylated both on the CapA1/CT-CapB2 kinase (lane 1) and on the DsRedEx-tag (lane 3). By contrast, tyrosine phosphorylation was detected on the DsRedEx-tag (lane 4) in the 4F mutant but not on the kinase moiety (lane 2), indicating that CapA1/CT-CapB2 phosphorylates the fused fluorescent protein tag when expressed in HEK293T cells. Autophosphorylation on the C-terminal tyrosine cluster does occur, but apparently is not required for kinase activity.

HEK293T cells were transfected with wild type or the 4F mutant LckN-DsRedEx-HRV3C-CapA1/CT-CapB2 plasmids. The expressed proteins were collected on anti-RFP magnetic beads and treated with HRV3C protease. CapA1/CT-CapB2 domains released by HRV3C treatment (lanes 1 and 2) and the DsRedEx-tags remaining bound to the beads (lanes 3 and 4) were analyzed by immunoblotting using anti-phosphotyrosine (PY), anti-CapB2, and anti-RFP (RFP) antibodies.
CapB2 still possessed activity after the deletion of 20 amino acids from the N-terminus (Fig. 3, lane 7), but phosphorylation significantly decreased by removal of 34 (Fig. 3, lane 8) residues. We also mutated other conserved amino acids in CapB2 that might be expected to be important for kinase activity and binding to CapA1. A G54A substitution in the Walker A, D79N substitution in the Walker A′, and D157N and P160A substitutions in the Walker B domains completely abrogated phosphorylation (Fig. 5, lanes 4, 7, 9, 10). These four residues, in addition to lysine 55, are evidently critical for kinase activity. Two other CapB2 mutants, D77N and R81H, apparently retained activity (Fig. 5, lanes 6 and 8). Although these two residues are highly conserved, they may not be absolutely required for the phosphotransfer reaction. The EX2RX2R motif (amino acids 23–29) is a conserved motif implicated in oligomerization of BY-kinases. Mutation of this region in E. coli Wzc decreased autophosphorylation and polysaccharide production23) but did not inhibit S. aureus CapB2 kinase activity.20) We changed the three conserved residues to one aspartic acid and two lysines (Fig. 5 lane 2, DKK) or to three alanines (lane 3, AAA) but did not notice any notable change in phosphotyrosine levels, suggesting that this motif is not essential for CapB2 kinase activity per se.

HEK293T cells were transfected with the indicated wild-type or mutant LckN-DsRedEx-CapA1/CT-CapB2 plasmids and subjected to immunoblotting using anti-phosphotyrosine (PY) and anti-RFP (RFP) antibodies.
It has been reported that CapA1 is a more efficient activator of CapB2 than CapA2.18,20) To test whether this observation could be reproduced in our mammalian expression system, we fused the C-terminal 28 amino acids of CapA2 to the N-terminus of full-length CapB2 to obtain CapA2/CT-CapB2. The two CapA/CT-CapB2 chimeras as well as CapB2 without the CapA activation domains were N-terminally-tagged with LckN-DsRedEx, transfected into HEK293T cells, and assessed for protein expression and tyrosine phosphorylation. As shown in Fig. 6, tyrosine phosphorylation of CapB2 (lane 1) clearly increased upon the fusion of CapA C-terminal cytoplasmic domains (lanes 2 and 3); however, we did not observe a clear difference between CapA1 and CapA2 in terms of CapB2 activation effect, since the phosphorylation levels of CapA1/CT-CapB2 (lane 2) and CapA2/CT-CapB2 (lane 3) were similar. Thus, CapA1 and CapA2 appeared to be equally potent CapB2 activators.

HEK293T cells were transfected with plasmids to express the indicated proteins fused to LckN-DsRedEx and subjected to immunoblotting using anti-phosphotyrosine (PY) and anti-RFP (RFP) antibodies.
Kinase activity has not been detected in CapB1 in spite of its being 70% similar to CapB2.18–20) Compared to the sequence from S. aureus subsp. aureus 6850 in the GenBank database, the CapB1 we cloned from our S. aureus strain had six amino acid differences (D117E, G136A, S139R, M146L, T196A, A213T) and a deletion of valine 127, which we will refer to as CapB1/Δ127V. CapB1/Δ127V was the only form isolated from the strain we used, and presumably is the sequence encoded in its genome.
For a closer comparison with previous reports, we restored the missing valine to obtain CapB1/127 V or introduced an isoleucine, which is the corresponding amino acid in CapB2, to obtain CapB1/127I. The three CapB1s were fused to the C-terminal activation domains of CapA1 and CapA2 to generate CapA1/CT-CapB1/Δ127V, CapA2/CT-CapB1/Δ127V, CapA1/CT-CapB1/127V, CapA2/CT-CapB1/127V, CapA1/CT-CapB1/127I, and CapA2/CT-CapB1/127I. As in the earlier experiments, the CapA/CT-CapB1 chimeras were N-terminally-tagged with LckN-DsRedEx and expressed in HEK293T cells.
Consistent with published results, tyrosine phosphorylation of CapA1/CT-CapB1/Δ127V or CapA2/CT-CapB1/Δ127V could not be detected (Fig. 6, lanes 5 and 6). Unexpectedly, however, CapA1/CT-CapB1/127V, CapA2/CT-CapB1/127V, CapA1/CT-CapB1/127I, and CapA2/CT-CapB1/127I were all tyrosine phosphorylated (lanes 8, 9, 11, 12), although to a lesser extent than CapA1/CT-Cap B2 and CapA2/CT-CapB2 (lanes 2 and 3). We checked for possible contamination of CapB2-expressing constructs in the plasmid preparations by PCR, but the results were negative (data not shown).
Similarly to CapB2, the kinase activities of CapB1/127V and CapB1/127I heavily depended on the CapA C-terminal activation domains. CapB1/127V and CapB1/127I without the activation domains were expressed in HEK293T cells (lanes 7 and 10), but no tyrosine phosphorylation could be detected, implying that CapB1 and CapB2 are activated through a similar mechanism.
To assess the importance of valine 127, we deleted the corresponding isoleucine 127 in CapB2 to produce CapB2/Δ127I. Phosphorylation of CapA1/CT-CapB2/Δ127I (Fig. 6, lane 14) was somewhat reduced compared with CapA1/CT-CapB2 but was still detectable, suggesting that the absence of valine 127 is not the only reason for the inactivity of CapB1/Δ127V. Replacement of the ATP-binding lysine with methionine in CapB1/127V and CapB1/127I abolished tyrosine phosphorylation (data not shown), supporting the notion that CapB1/127V and CapB1/127I fused to CapA C-terminal tails does possess tyrosine kinase activity, at least when expressed in HEK293T cells.
The activities of BY-kinases have been measured by using purified enzymes and 32P-ATP. Although radioisotopic detection is still considered the gold standard assay for protein kinases, we believe that more convenient methods would contribute to enzyme characterization and inhibitor development of BY-kinases. If BY-kinases could be expressed in mammalian cell lines and their activity detected by immunoblotting, this would also provide a convenient and useful means of investigating residues and domains required for enzymatic function.
S. aureus CapB2 is one of the best characterized BY-kinase to date. We investigated whether this bacterial protein can be expressed as an active tyrosine kinase in HEK293T cells. We attached various tags to the CapA1/CT-CapB2 protein and examined the levels of both protein and phosphotyrosine. CapA1/CT-CapB2 translated into active kinases only when fluorescent proteins were fused on the N-terminus. GST- and His-tagged CapB2 kinases have been successfully expressed in E. coli, but all our attempts to express BY-kinases with nonfluorescent protein tags in HEK293T cells, including others not shown here, have failed thus far. Presumably, in human cells, BY-kinases are unstable and cannot be expressed unless stabilized by fusion to fluorescent proteins that have rigid structures.
It has been proposed that nonphosphorylated CapA1/CT-CapB forms an inactive octamer that dissociates into active monomers upon phosphorylation of the C-terminal tyrosine cluster.19) Reversible phosphorylation of the C-terminus may regulate monomer–oligomer conversion, which in turn controls kinase activity and hence various biological functions of CapB2. In our experiments, however, phosphorylation of the C-terminal tyrosine cluster did not appear to affect kinase activity of CapA1/CT-CapB2. Tyrosine phosphorylation was not compromised in the mutant whose C-terminal tyrosines were converted to phenylalanines. CapA1/CT-CapB2 expressed in HEK293T cells phosphorylated not only itself but also the attached fluorescent protein tag.
These characteristics of CapB2 expressed in HEK293T cells resemble those of the B. subtilis PtkA.11) Autophosphorylation did not affect kinase activity and, even when all the C-terminal tyrosines were replaced with phenylalanines, PtkA still phosphorylated GST and maltose binding protein fused to its activator PtkB. BY-kinases have been hypothesized to possess relaxed substrate specificity.4) We have observed that CapB2 is capable of phosphorylating various fluorescent proteins other than DsRedEx, and it therefore seems plausible that CapB2 is a promiscuous tyrosine kinase that phosphorylates various tyrosine residues in its vicinity.
In experiments using purified proteins, CapB2 was more efficiently activated by CapA1 than its cognate effector, CapA2.18,20) However, in our mammalian cell expression system, CapA1 and CapA2 activated CapB2 kinase comparably. It has been shown that the penultimate phenylalanine of CapA1 directly interacts with the adenine moiety of ATP19) and mutation of this phenylalanine to alanine decreased autophosphorylation of CapA1/CT-CapB2. The CapA and CapB sequences we used are those of S. aureus ssp. aureus 6850. In this strain, the second-last amino acid of CapA1 is tyrosine 204, and not phenylalanine. We thought that this could be the reason for the difference and mutated this tyrosine to phenylalanine. However, we did not observe a significant difference in tyrosine phosphorylation between the tyrosine and the phenylalanine versions (data not shown).
CapB1 shares more than 70% homology with CapB2, but its kinase activity has never been demonstrated. Despite comprehensive biochemical and physicochemical studies, the reason for its kinase inactivity has not been clearly explained.20) The CapB1 we cloned was also inactive but had six amino acid changes and a deletion of valine 127 compared with the sequence in the data base. None of the six amino acid changes occurred in a conserved residue, and so we speculated that they did not affect kinase activity. We were somewhat concerned about the deletion, however, and to confirm that CapB1 lacks kinase activity, we complemented the missing valine 127. Unexpectedly, insertion of valine or isoleucine apparently converted the catalytically inert protein into an active tyrosine kinase. On the other hand, deletion of the corresponding isoleucine 127 in CapB2 decreased, but did not completely abolish, kinase activity. Again, the reason for this discrepancy with previous publications is unclear, although it may have resulted from differences in the attached N-terminal tags or the hosts used for protein expression.
The valine 127 position lies between the fourth alpha-helix and the seventh beta-sheet of CapB1.20) The region surrounding this amino acid has not been recognized as a signature motif but is conserved throughout BY-kinases, including those of Gram-negative bacteria, and so could be viewed as another common feature of BY-kinases.4) We speculate that amino acid 127 and its neighborhood, in combination with other regions, play some role in CapB kinase activity or interaction with CapA. The CapB1/Δ127V we originally cloned did not exhibit detectable kinase activity, and whether CapB1 actually functions as a kinase in bacteria remains to be investigated.
BY-kinases function in concert with many other factors, and results obtained from a heterologous expression system may not provide much useful information regarding their physiological roles in bacteria. Nevertheless, we feel that the mammalian expression system described here offers various advantages. Most notably, kinase activity can be analyzed without protein purification. In addition, since the transfected kinase is the only bacterial protein in the cells, mutants can be analyzed without interference from endogenous wild-type enzymes. Over the course of this study, we created more than 70 BY-kinase constructs, examined their tyrosine kinase activities, and revealed several regions and residues important for kinase activity. This would have been a much more labor-intensive and time-consuming process had it been done using isotopic protein kinase assays and purified enzymes. Although the data obtained must be interpreted with caution, we anticipate that the method described here could be applied to the characterization of BY-kinases from other bacteria as well as to inhibitor screening.
This study was supported by a Grant-in-Aid for Scientific Research from The Japan Society for Promotion of Science.
The authors declare no conflict of interest.