Abstract
Oxyresveratrol (ORV) is a naturally extracted compound with many pharmacological activities. However, information about the crystalline form is not known when considering the development of a form for oral dosage. Cocrystal engineering offers drug molecular understanding and drug solubility improvements. Thus, we attempted cocrystallization of ORV using 10 carboxylic acids as a coformer at a 1:1 M ratio. Each combination was processed with liquid-assisted grinding, solvent evaporation and a slurry method, then characterized by powder X-ray powder diffraction (PXRD), conventional and low-frequency Raman spectroscopy and thermal analysis. The solubility, dissolution and permeation studies across Caco-2 cell monolayers were conducted to evaluate the ORV samples. A screening study revealed that an ORV and citric acid (CTA) cocrystal formed by ethyl acetate-assisted grinding had characteristic PXRD peaks (14.0 and 16.5°) compared to those of ORV dihydrate used as a starting material. Low-frequency Raman measurements, with peaks at 100 cm−1, distinguished potential cocrystals among three processing methods while conventional Raman could not. An endothermic melt (142.2°C) confirmed the formation of the novel crystalline complex. The solubility of the cocrystal in the dissolution media of pH 1.2 and 6.8 was approximately 1000 µg/mL, a 1.3-fold increase compared to ORV alone. In vitro cytotoxicity studies showed that the cocrystal and physical blend were not toxic at concentrations of 25 and 12.5 µM ORV, respectively. The ORV-CTA cocrystal enhanced the cellular transport of ORV across Caco-2 monolayers. Therefore, cocrystallization could be used to improve aqueous solubility and permeability, leading to better oral bioavailability of ORV.
INTRODUCTION
Oxyresveratrol (ORV) is a polyphenolic stilbene found in the heartwood of the plant Artocarpus lakoocha ROXBURGH (Moraceae). ORV has been used in Thai traditional medicine1) and is reported to have many pharmacological activities, namely, antioxidant/anti-inflammatory activities,2,3) tyrosinase enzyme inhibitory activities4) and strong neuroprotective activities.5) ORV also possesses antiviral activities against herpes simplex virus (HSV-1 and HSV-2),6,7) varicella zoster virus8) and influenza virus.9) However, information on the crystalline form is unknown for the development of the oral dosage form.
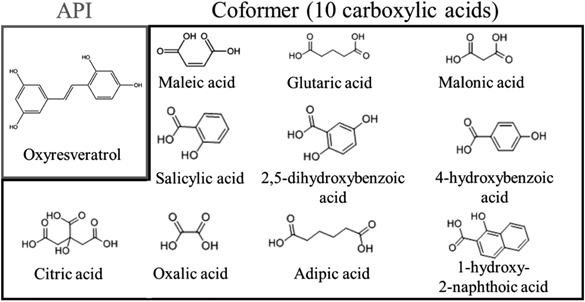
In recent years, exploring the crystalline form of drug substances, including crystalline polymorph, amorphous, salt, cocrystal and solvate,10) is typically performed at an early stage of new drug development.11) Among them, the design of pharmaceutical cocrystals is an attractive technique as it offers a number of advantages, for example, understanding the drug molecular structure and intermolecular bonding, and in particular improving physicochemical properties of active pharmaceutical ingredients (APIs) such as drug solubility and dissolution rates.12,13) A cocrystal (Fig. 1) is a crystalline complex that consists of a drug substance and a cocrystal former (coformer). The cocrystal from such neutral molecules is formed through weak intermolecular interactions such as hydrogen bonding.14) In contrast, a salt is often used to improve solubility, and cannot be formed without a dissociating group subject to acid/base reaction. In addition, the properties of the coformer can reflect the physicochemical properties of the cocrystal; thus, it is possible to improve the solubility and stability of the API without changing its chemical structure and therapeutic effect.15) Carboxylic acids are commonly used as coformers in pharmaceutical additives.14) Therefore, we studied the characterization of ORV and cocrystalline formation using a variety of carboxylic acids (Fig. 2).
Screening methods are widely used for examining combinations of API and various coformers to search for new cocrystals. A mix grinding method with a mechanochemical reaction has been reported as a promising method with high quality and high yield of cocrystals.16) Mechanochemistry of organic materials has been applied to multicomponent cocrystallization processing of biological materials, and has then led to the discovery of numerous substances that cannot be produced by conventional solution-based approaches.17) In liquid assisted grinding (LAG), samples are ground with a trace amount of organic solvent, which plays an important role in cocrystal formation.18) While processing via solvent evaporation (SE), the solution of molecules is expected to undergo various hydrogen bonding reactions to form cocrystals that produce lower energy and are more homogeneous in terms of crystal composition.19) In suspension via the slurry method (Slu), the API and coformer are also expected to form via hydrogen bonding. The slurry technique is highly efficient for cocrystal screening with the advantage over other traditional methods that cocrystal formation is not limited to noncongruent solubility of cocrystal components and has a flexible range in terms of physical stability of the phase diagram and solvent selection approaches based on thermodynamically driven solution-mediated phase transformation.20–24)
Compared to cocrystal physicochemical characterizations, permeation studies of cocrystal have been limited even though membrane permeability is one of the most important characteristics determining drug performance during absorption or bioavailability. The cocrystals were to be non-toxic at concentrations up to 1 mM in in vitro cytotoxicity studies using Calu-3 cells and had better drug permeability than the drug alone by changing the efflux effect.25) On the other hand, using the permeation apparatus with cellulose membrane, the enhanced dissolution and permeation properties of the cocrystals tested in the form of a powdered drug substance with and without polymers,26) polymorphic cocrystals conducted in pH 1.2 and 7.4 solutions,27,28) cocrystals with various coformers at a 1 : 1 ratio in phosphate buffer29) and biologically active cocrystals stabilized via a certain lattice energy30) have been revealed.
In this study, we attempted to increase the possibility of new cocrystalline formation in a screening test by using three different methods (LAG, SE and Slu) cocrystallized with 10 coformers (Fig. 2). All resultant samples were characterized and confirmed by a series of analytical tools. Low-frequency Raman spectroscopy was applied to identify the crystal form and was investigated for its usefulness of cocrystal identification. The dissolution test based on Japanese pharmacopeia was conducted to evaluate an improvement in the dissolution behavior of ORV. We also evaluated the cytotoxicity and permeability of the cocrystal of ORV (SE) using Caco-2 cell monolayers as an in vitro intestinal absorption model, in comparison with a physical mixture of ORV (PM). To our knowledge, the use of Caco-2 cells for the prediction and evaluation of cellular uptake and transport of cocrystal-alone has not been reported. We hereby first established the use of Caco-2 cells as a gastric and intestinal barrier to predict the absorption of cocrystallized natural products.
MATERIALS AND METHODS
MaterialsORV was obtained from the Department of Pharmacognosy and Pharmaceutical Botany, Faculty of Pharmaceutical Sciences, Chulalongkorn University (Bangkok, Thailand). Maleic acid (MALE), adipic acid (ADP), malonic acid (MALO), 2,5-dihydroxybenzoic acid (2,5-DBA), oxalic acid (OXA), 1-hydroxy-2-naphthoic acid (1-H-2-BA), salicylic acid (SAL) and citric acid (CTA) were purchased from Wako Pure Chemical Corporation (Osaka, Japan). 4-Hydroxybenzoic acid (4-HBA) and glutaric acid (GLU) were purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). ORV was used as the API, and the others were used as coformers. Ethyl acetate, chloroform, sodium dihydrogen phosphate, anhydrous disodium hydrogen phosphate, sodium chloride and hydrochloric acid were purchased from Wako Pure Chemical Corporation. 3-[4,5-Dimethyltiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) was purchased from Merck Millipore (Billerica, MA, U.S.A.). Dulbecco’s modified Eagle’s medium (DMEM), L-glutamine, nonessential amino acids, penicillin and streptomycin, and fungizone were obtained from Invitrogen (Grand Island, NY, U.S.A.). Fetal bovine serum (FBS) was purchased from PAA Laboratories (Haidmannweg, Austria).
Preparation of CocrystalsORV and 10 carboxylic acids were weighed to a total 100 mg at a molar ratio of 1 : 1 and mixed in an agate mortar to obtain physical mixtures (PM). Each combination was then mixed in a 2.0 mL sample tube with two grinding balls (5 mm diameter) and 20 µL solvent was added (ethyl acetate and chloroform). To prepare a liquid assisted grinding mixture (LAG), the PM were ground for 24 h using a multisample vibrating ball mill (MSC-100, BMS, Tokyo, Japan) under a rotational speed of 1500 rpm and a temperature of 60°C. For the SE method, PM were dissolved in ethyl acetate and the solvent was evaporated under reduced pressure. For the slurry (Slu) method, PM were suspended in ethyl acetate, stirred and suction filtered.
Characterizations of the Crystalline FormORV, coformer, PM, LAG, SE and Slu samples were characterized by the following analytical techniques:
Powder X-Ray Diffraction (PXRD) MeasurementPXRD patterns were carried out using a powder X-ray diffractometer (MiniFlex 600, Rigaku, Tokyo, Japan). The sample (50 mg) was spread on a glass plate. The diffraction angle, the scan rate, the voltage and the current of the generator were set to 5.0 to 35.0°, 4.0°/min, 40 kV and 15 mA, respectively.
Thermogravimetry-Differential Thermal Analysis (TG-DTA) MeasurementTG-DTA experiments were conducted on a differential thermal analyzer (Thermo plus EVO 2 TG 8121, Rigaku) using nitrogen gas as dry air with a flow rate of 100 mL/min. Approximately 5 mg of sample was placed in open aluminum pans. The sample was heated from 25 to 250°C at a rate of 10°C/min.
Conventional and Low-Frequency Raman Spectroscopy MeasurementFor measurement of the conventional region (200 to 2400 cm−1), a microscopic Raman spectrometer (Workstation, Kaiser Optical Systems Inc., CA, U.S.A.) was used. The laser wavelength was 785 nm, exposure time was 5 s, and accumulation count was 3 times. A low wavenumber Raman spectroscopy unit (THz-Raman Micro, Ondax Inc., MI, U.S.A.) was used for measurement in the low wavenumber region (−200 to 200 cm−1). The Raman spectra were measured under a laser wavelength of 976 nm, exposure time of 5 s, and accumulation count of 3 times.
Dissolution TestTo evaluate the improvement in the physical properties, a dissolution test was performed with ORV and a combination of ORV and CTA. Approximately 1 g of ORV powder (or corresponding to 1 g of ORV powder, for the crystalline complex) was weighed. JP 1 (pH 1.2) and JP 2 (pH 6.8) (300 mL) in the Japanese Pharmacopoeia were used as dissolution medium. The dissolution tests were performed with a rotational speed of 100 rpm at 37 ± 0.5°C by a paddle method with the dissolution tester (NTR-3000, Toyama Industry Co., Ltd., Osaka, Japan). At 5, 10, 15, 20, 25, 30, 60, 90, and 120 min, 5 mL of the sample was removed and replaced. The absorbance at 325 nm of the sample solution was measured using an UV-visible spectrophotometer (UVmini-1240, Shimadzu, Kyoto, Japan), and the concentration of ORV at each time point was calculated.
Cell CultureThe human colon adenocarcinoma cell line, Caco-2, was obtained from the American Type Culture Collection (ATC C HTB37, Rockville, MD, U.S.A.). The cells were cultured in a complete medium (cDMEM) containing 15% (v/v) heat-inactivated fetal bovine serum (FBS), 1% (v/v) L-glutamine, 1% (v/v) nonessential amino acids, 1% (v/v) penicillin-streptomycin (antibiotic), 0.2% (v/v) fungizone (antifungal) and serum free media (DMEM) at 37°C in a humidified atmosphere with 5% CO2.
Evaluation of ORV Permeation across Caco-2 MonolayersCytotoxicity StudiesThe cytotoxicity of SE and PM samples to Caco-2 cells was determined using an MTT assay. The Caco-2 cells were seeded in 96 well plates at a density of 1 × 104 cells/mL in 200 µL cDMEM and incubated at 37°C in an atmosphere with 5% CO2 and 95% relative humidity for 24 h. The old medium was removed, and cells were washed with DMEM. The cells were treated with SE or PM at equivalent concentrations of ORV (12.5, 25, 50, 100, or 200 µM in DMEM) for 24 h. Cells treated with DMEM were used as a control group. After treatment, 20 µL of MTT solution (5 mg/mL in phosphate buffered saline (PBS)) was added to each well and incubated for another 4 h (37°C, 5% CO2). Finally, the medium was removed, and 200 µL of dimethyl sulfoxide (DMSO) was added to dissolve the formazan crystals. The absorbance of formazan was measured at 540 nm by a microplate reader (SPECTROstar, BMG LABTECH, Germany). The experiment was performed in four replicates. The concentrations of SE and PM that produced cell viability that was not significantly different from the control were chosen for further evaluation of cellular transport.
Evaluation of Cellular TransportThe cellular transport of the SE and the PM samples was determined using the Caco-2 cell monolayers model. Caco-2 cells with passages between 25 and 35 were used. The Caco-2 cells were seeded in 6 well trans-well membrane inserts (diameter 24 mm, pore size 0.4 µm; ThinCerts™-TC Einsatze, Greiner Bio-One, Switzerland) at a density of 4 × 104 cells/well with 2 mL of cDMEM in both the apical and basolateral compartments. The cell cultures were maintained at 37°C under 95% humidity and 5% CO2. The serum (FBS) content of the complete medium was decreased to 7.5% once the cultures reached confluence. The complete medium was changed every other day. At 21–24 d after confluence with a transepithelial electrical resistance (TEER) between 500–700 Ω/cm2, differentiated monolayers were washed with serum free medium before adding 2 mL of freshly prepared serum free medium containing the SE or the PM samples with a nontoxic concentration at 12.5 µM equivalent of ORV. The basolateral compartment filled with 2 mL of phenol red-free DMEM. The medium in the apical compartment, cell pellet and basolateral compartment after the sample treatment were collected at 15, 30, 60, 90, and 120 min. The experiments were performed in 3 replicates. All samples were blanked with nitrogen gas and stored at −80°C for further experiments. The apparent permeability coefficients (Papp, cm/s) were calculated according to the following equation:
where dQ/dt is the permeability rate (µM/s), V is the volume of the basolateral chamber (2 cm3), A is the surface area of the insert (4.524 cm2), and C0 is the initial concentration (µM) of the samples.
Extraction of ORV from Bioavailable Fractions for HPLC AnalysisBriefly, 2 mL of each sample was transferred to a 15-mL centrifuge tube and mixed with 2 mL of 0.1 M pH 7.1 potassium phosphate buffer containing 100 U of β-glucuronidase and 0.1 U of sulfatase. The mixture was incubated in a shaking water bath for 3 h at 37°C. Then, 1 mL of 1% sodium dodecyl sulfate (SDS) in ethanol was added and subsequently vortexed for 1 min. Ethyl acetate at a ratio of 1 : 1 was added to the mixture, sonicated for 10 min, vortexed for 2 min and centrifuged (ROTINA 380 R, Germany) at 4000 × g for 10 min. The organic layer was collected into a 10-mL vial. The extraction was repeated twice, and the organic layers were combined and dried under nitrogen gas blowing. The residue was reconstituted with a mobile phase prior to HPLC analysis.
Chromatographic ConditionsAll samples were analyzed on an Agilent series 1290 UHPLC (Agilent Technologies, Palo Alto, CA, U.S.A.) liquid chromatograph equipped with binary pump, autosampler, thermostat column compartment, degasser, and diode array detector (DAD). The UHPLC system was operated with an Agilent ChemStation B 3.0 software. A HALO C18 column (4.6 × 100 mm, 5 µm) was used for separation and was maintained at 25°C. The injection volumes were 5 µL of the samples. The mobile phase consisted of 0.1% aqueous formic acid and methanol (55 : 45, v/v) and was delivered at 1 mL/min. The detection of ORV was monitored at 320 nm.
RESULTS AND DISCUSSION
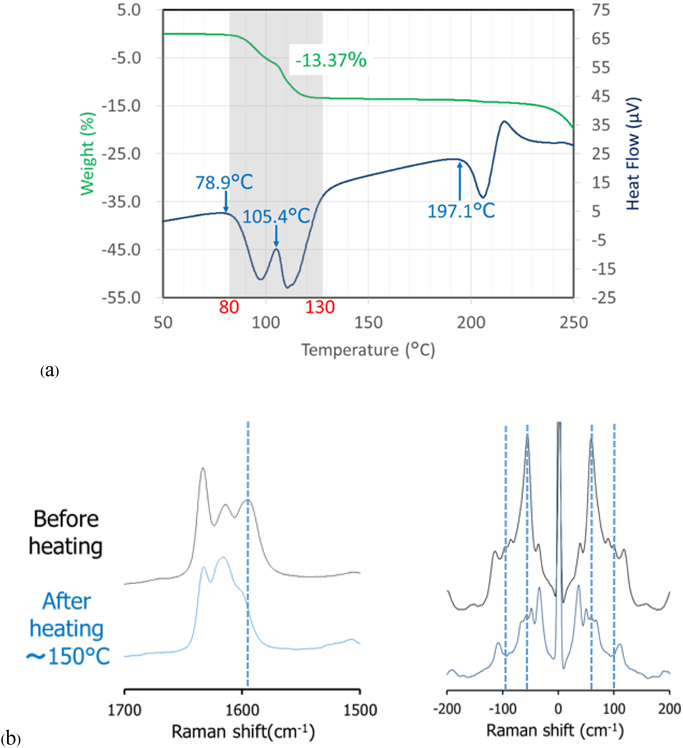
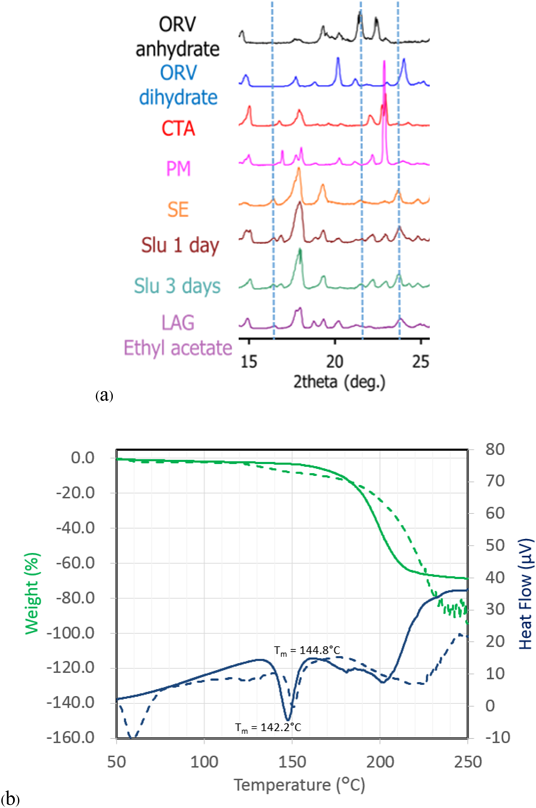
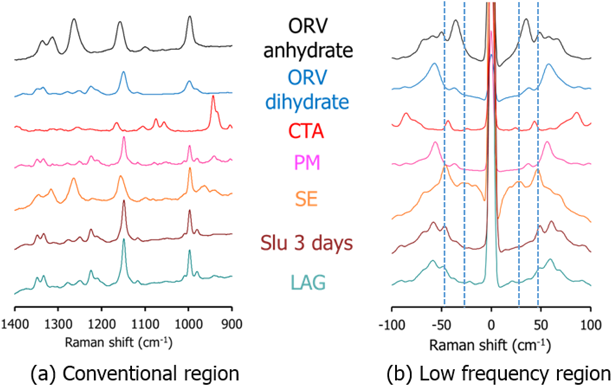
Characterization of ORVThe TG-DTA curves of ORV (Fig. 3a) show a step change of 13.37% weight loss along with 2 endothermic events from 80 to 130°C indicating a 1 : 2 M ratio of ORV : H2O. Consequently, the starting material of ORV could be dihydrate. The following 200°C endothermic peak of ORV anhydrate melt was followed by an exothermic peak, possibly ascribed to crystallization of ORV anhydrate. The decomposition occurred at 260°C, as seen from the sharp weight change in the TG trace. Supplementary Fig. S1 demonstrates changes in the PXRD pattern before and after heating the ORV. Instead of characteristic diffraction peaks at 20.2, 21.2 and 23.0°,1) heating ORV to 150°C could transform ORV to its anhydrous form showing peaks at 19.3, 21.5 and 22.4°. This observation is because ORV dihydrate becomes anhydrate with heat. In terms of the Raman spectra (Fig. 3b), after heating, a peak at 1590 cm−1 disappeared in the conventional region, whereas in the low frequency region, a large sharp peak at 50 cm−1 decreased, and the small peak at 100 cm−1 disappeared. Thus, the conversion of ORV dihydrate to the anhydrate form could be confirmed and distinguished by the Raman spectra with the TG-DTA profiles and PXRD patterns.
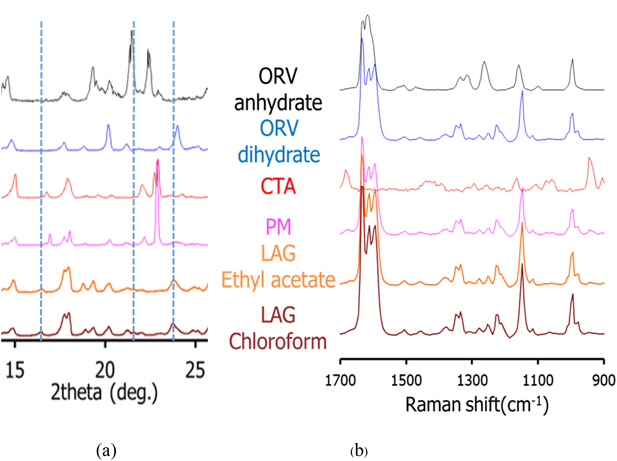
Cocrystal Exploration of ORVTable 1 summarizes a screening result of cocrystallization for ORV using 10 coformers characterized by PXRD. Characteristic diffraction peaks of potential cocrystals were observed in the samples processed with the LAG method, with the combination of ORV and GLU or CTA. In this paper, we mainly focus on the combination of ORV and CTA with their specific properties. Characteristic peaks appeared at 14.0 and 16.5° in ORV-CTA samples processed with the LAG method (Fig. 4a, Supplementary Fig. S2). The PXRD patterns from the ethyl acetate and chloroform assisted samples were in good agreement and different from starting materials. On the other hand, in the conventional region Raman spectra, there was no obvious change in the processed samples compared to the PM of ORV-CTA (Fig. 4b). It is interesting that the changes in PXRD patterns of the LAG samples were observed while no changes were detected using conventional Raman measurements. This finding might be because partial cocrystal was formed by LAG and conventional Raman is not very sensitive or specific for such identification, and generally gives information on the functional groups.31)
Table 1. Evaluation of Cocrystallization by PXRD
| Coformer | Cocrystallization or NOT |
|---|
| Added solvent |
|---|
| Ethyl acetate | Chloroform |
|---|
| Maleic acid | × | × |
| Adipic acid | × | × |
| Malonic acid | × | × |
| 2,5-Dihydroxybenzoic acid | × | × |
| Oxalic acid | × | × |
| 4-Hydroxybenzoic acid | × | × |
| 1-Hydroxy-2-naphthoic acid | × | × |
| Glutaric acid | ○ | × |
| Salicylic acid | × | × |
| Citric acid | ○ | ○ |
○: potential ×: no potential.
For comparison of the different production methods, changes in the PXRD patterns and Raman spectra of SE, Slu and LAG are shown in Figs. 5a and 6 and Supplementary Fig. S3, respectively. Peaks at 14.0 and 16.5° agree well for the SE, Slu and LAG samples. Notably, SE illustrates clearer PXRD peaks than LAG and Slu, indicating that the SE method tends to be more appropriate for more complete formation than LAG and Slu, which have partial cocrystal formation. In the conventional region Raman measurement, the SE spectrum seems to be similar to ORV anhydrate with no characteristic peaks while in the low frequency region, characteristic spectra with the specific peaks at 35.1 and 50 cm−1 and without the peak approximately 40 cm−1 can differentiate processed samples (SE, Slu and LAG) from starting materials (Fig. 6). Here, it is apparent that the low-frequency Raman is more advantageous and suitable for determination of cocrystallization.32)
The TG-DTA curve is shown in Fig. 5b. The melting temperature (Tm) of ORV-CTA (SE) and the coformer (CTA) is 142.2 and 144.8°C, respectively. It is clear that the endothermic peak of ORV-CTA shifts to a lower temperature compared to the starting materials (Tm of ORV = 197.1°C). Thus, a novel crystalline complex was formed between ORV and CTA.
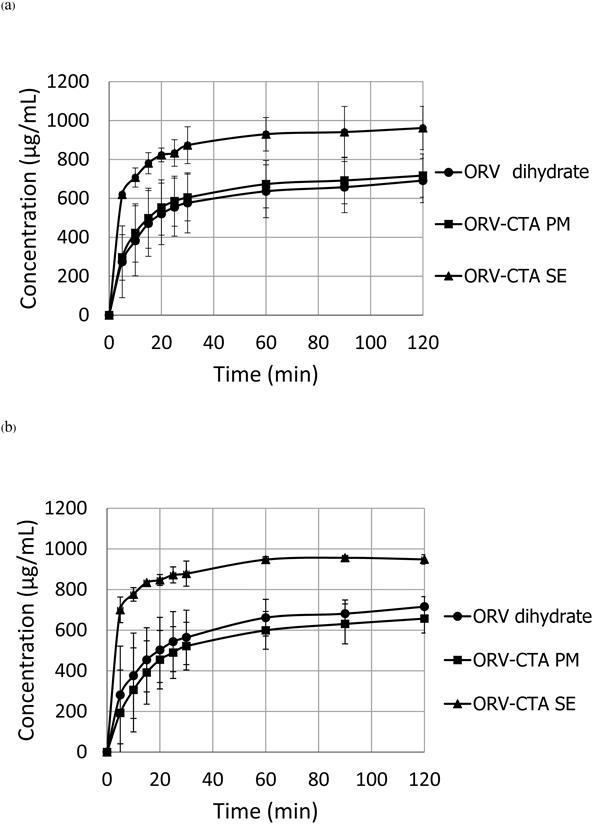
Dissolution Tests of the ORV CocrystalDissolution tests of ORV, the ORV-CTA physical mixture and the crystalline complex were conducted in physiological solutions, including acid and phosphate buffer stages, as shown in Fig. 7. The cocrystal has solubility in pH 1.2 and 6.8 media of approximately 1000 µg/mL, whereas that of ORV and its PM were approximately 750 µg/mL. These findings confirmed the improved solubility of the ORV complex. In addition, there was no difference in dissolution profiles due to the pH change. This result means the obtained complex is cocrystal, not salt, because ORV is a neutral compound that behaves similarly in the acid-buffer condition. The cocrystal has smaller pKa value than the salt, resulting in no difference in dissolution profiles upon a pH change.33) If a charged compound formed a salt, it would be affected by the pH change.
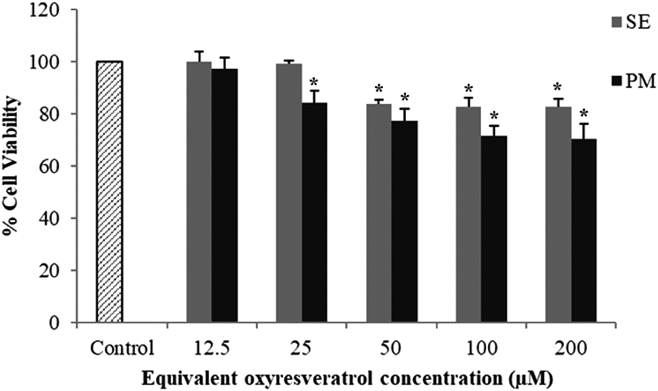
Permeability of ORV Transport across Caco-2 Cell MonolayersPrior to the cellular transport study, the evaluation of the cytotoxicity of SE and PM samples was performed on Caco-2 cells and compared to the control group. Increasing the ORV concentration could reduce the % cell viability significantly (p < 0.05) (Fig. 8). However, the maximum nontoxic concentrations of the SE and PM samples were observed at 25 and 12.5 µM, respectively. Therefore, the 12.5 µM concentration of SE and PM in contact with the cells in the cellular transport study was chosen for subsequent permeability studies.
In the cellular transport study of a drug using Caco-2 model, the monolayer integrity of the cells should be evaluated to ensure that the model can represent the gastrointestinal epithelial permeability barrier. There are several methods to assess the integrity of the monolayer including trans-epithelial electrical resistance (TEER) measurement, phenol red assay and [14C] mannitol assay.34,35) The TEER measurement and phenol red assays are widely used in the evaluation of the monolayer integrity.36–39) since they are simple and non-radioactive. In this study, we therefore used the TEER measurement and phenol red assay to evaluate the monolayer integrity before and during the cellular transport experiment. The phenol red-containing DMEM was used as a medium on the apical compartment while the phenol red-free DMEM was added on the basolateral side to maintain pH, osmotic and nutrient balance to the cells. In addition, the use of the phenol red-free DMEM on the basolateral side allowed us to monitor the passage of phenol red across the Caco-2 monolayer due to monolayer leakage caused by the test compounds. In this experiment, no phenol red was observed in the basolateral side suggesting the integrity of the monolayer during the transport of ORV.
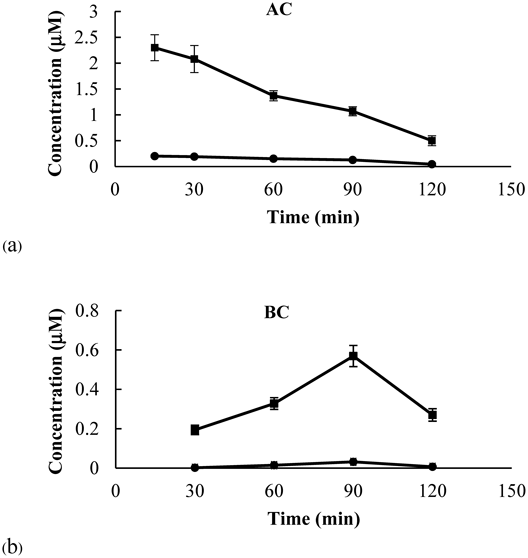
The amount of ORV from the SE and PM samples in the apical compartment (AC) decreased in a time-dependent manner (Fig. 9a). The amount of ORV from the SE sample was noticeably greater than that from the PM sample at all time points. This result could be the consequence of ORV cocrystallization in terms of enhanced solubility. Surprisingly, in the basolateral compartment (BC), at 30 min, the amount of ORV in the PM-treated group was 0.004 µM whereas in the SE-treated group the amount of ORV was 0.194 µM, which was approximately 50 times greater than that of the PM-treated group (Fig. 9b). In addition, Fig. 9b illustrated that the amount of ORV in the BC of both groups increased over time and reached the maximum at 90 min. At 90 min, the concentration of ORV in the SE-treated group (0.569 µM) was 18 times greater than that of the PM-treated group (0.032 µM).
The concentrations of ORV found on the BC side accounts for the transport of ORV from AC to BC, transport of ORV from BC to AC, and the chemical degradation of ORV in the acceptor medium. We postulated that the rate of ORV transport from AC to BC during 0–90 min was higher than the rate of ORV transport from BC to AC and the degradation rate of ORV in the medium. However, the degradation rate of ORV in the medium after 90 min would become predominant because the amount of ORV on the AC side decreased by ca. 50% and the half-life of ORV in DMEM was 59 min40) resulting in the reduction of the concentration on the BC side. This finding was similar to that observed in resveratrol and another stilbenoid.41)
Nevertheless, for both SE and PM samples, the increasing amount of ORV in the BC were strongly correlated to the decreasing amounts of ORV dissolved in the AC. The apparent permeability coefficient (Papp) value of ORV from AC-to-BC was calculated after incubating the Caco-2 cells with SE and PM for 30 min to 90 min. The Papp of SE [(3.68 ± 0.21) × 10−6 cm/s] was significantly greater than the value of PM [(0.30 ± 0.01) × 10−6 cm/s], suggesting that the ORV transport from the SE sample was greater than that from the PM sample. It was possible that the CTA coformer played an important role in changing the flux rate of ORV due to modification of the structure and intermolecular bonding of API by cocrystallization.
The high solubility of the coformer tends to result in higher solubility of cocrystals; on the contrary, high lipophilicity with a log P value greater than the parent molecule was reported to increase membrane permeation.28,42) In the case of ORV-CTA, the log P value of CTA (−1.7) was much lower than that of ORV (2.8), the trade-off between solubility and log P that can lead to opposing effects on the overall permeability enhancement was mainly due to the solubility of API and exposure time interval. Herein, only the dissolved (dissociated from cocrystal) API can completely permeate the membrane over time. On the other hand, a decrease in the melting temperature of the cocrystal, corresponding to the mp. of ORV (197.1°C) and ORV-CTA (SE) (142.2°C), can also affect solubility, and hence its permeability, as revealed by Yan et al.42) Additionally, structural arrangements, motions of molecules and the van der Waals interactions of the compounds can have effects on the cell transport properties.28)
CONCLUSION
A novel pharmaceutical cocrystal of a multipurpose drug, ORV, with a coformer (CTA) can be formed from the screening of 10 carboxylic acids through a classic solvent evaporation method. The formation of such crystalline complexes was confirmed by PXRD studies and other auxiliary tools including Raman spectroscopy and TG-DTA. In particular, low-frequency Raman can differentiate the cocrystal by scattering peaks that were not detected by conventional Raman. Hence, the low-frequency Raman measurement is superior in cocrystal identification. The ORV-CTA cocrystal has a 1.3-fold increase in solubility and relative dissolution profiles in both pH 1.2 and 6.8 media, implying cocrystal formation rather than formation of its salt. Permeation studies using Caco-2 cell monolayers emphasized the enhanced permeability of the cocrystal. In conclusion, cocrystallization of ORV-CTA was successfully produced as a new chemical compound with improved aqueous solubility and permeability across the human intestinal tract. The cocrystal formation could lead to better drug absorption and oral bioavailability of ORV.
Acknowledgments
We thank the Asia Africa Center for Drug Discovery at Meiji Pharmaceutical University (MPU-AACDD), Japan; Research Affairs and Natural Products for Ageing and Chronic Diseases Research Unit, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Thailand to support funding to this research. This work was also supported in part by JSPS KAKENHI, a Grant-in-Aid for the Scientific Research (C), Grant Number 17K08253 (to T.F.).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Sangsen Y, Wiwattanawongsa K, Likhitwitayawuid K, Sritularak B, Wiwattanapatapee R. Modification of oral absorption of oxyresveratrol using lipid based nanoparticles. Colloids Surf. B Biointerfaces, 131, 182–190 (2015).
- 2) Chung KO, Kim BY, Lee MH, Kim YR, Chung HY, Park JH, Moon JO. In-vitro and in-vivo anti-inflammatory effect of oxyresveratrol from Morus alba L. J. Pharm. Pharmacol., 55, 1695–1700 (2003).
- 3) Lorenz P, Roychowdhury S, Engelmann M, Wolf G, Horn TF. Oxyresveratrol and resveratrol are potent antioxidants and free radical scavengers: effect on nitrosative and oxidative stress derived from microglial cells. Nitric Oxide, 9, 64–76 (2003).
- 4) Tengamnuay P, Pengrungruangwong K, Pheansri I, Likhitwitayawuid K. Artocarpus lakoocha heartwood extract as a novel cosmetic ingredient: evaluation of the in vitro anti-tyrosinase and in vivo skin whitening activities. Int. J. Cosmet. Sci., 28, 269–276 (2006).
- 5) Weber JT, Lamont M, Chibrikova L, Fekkes D, Vlug AS, Lorenz P, Kreutzmann P, Slemmer JE. Potential neuroprotective effects of oxyresveratrol against traumatic injury. Eur. J. Pharmacol., 680, 55–62 (2012).
- 6) Likhitwitayawuid K, Sritularak B, Benchanak K, Lipipun V, Mathew J, Schinazi RF. Phenolics with antiviral activity from Millettia erythrocalyx and Artocarpus lakoocha. Nat. Prod. Res., 19, 177–182 (2005).
- 7) Lipipun V, Sasivimolphan P, Yoshida Y, Daikoku T, Sritularak B, Ritthidej G, Likhitwitayawuid K, Pramyothin P, Hattori M, Shiraki K. Topical cream-based oxyresveratrol in the treatment of cutaneous HSV-1 infection in mice. Antiviral Res., 91, 154–160 (2011).
- 8) Sasivimolphan P, Lipipun V, Likhitwitayawuid K, Takemoto M, Pramyothin P, Hattori M, Shiraki K. Inhibitory activity of oxyresveratrol on wild-type and drug-resistant varicella-zoster virus replication in vitro. Antiviral Res., 84, 95–97 (2009).
- 9) Liu AL, Yang F, Zhu M, Zhou D, Lin M, Lee SM, Wang YT, Du GH. In vitro anti-influenza viral activities of stilbenoids from the lianas of Gnetum pendulum. Planta Med., 76, 1874–1876 (2010).
- 10) Aitipamula S, Banerjee R, Bansal AK, Biradha K, Cheney ML, Choudhury AR, Desiraju GR, Dikundwar AG, Dubey R, Duggirala N, Ghogale PP, Ghosh S, Goswami PK, Goud NR, Jetti RRKR, Karpinski P, Kaushik P, Kumar D, Kumar V, Moulton B, Mukherjee A, Mukherjee G, Myerson AS, Puri V, Ramanan A, Rajamannar T, Reddy CM, Rodriguez-Hornedo N, Rogers RD, Row TNG, Sanphui P, Shan N, Shete G, Singh A, Sun CC, Swift JA, Thaimattam R, Thakur TS, Kumar Thaper R, Thomas SP, Tothadi S, Vangala VR, Variankaval N, Vishweshwar P, Weyna DR, Zaworotko MJ. Polymorphs, salts, and cocrystals: what’s in a name? Cryst. Growth Des., 12, 2147–2152 (2012).
- 11) Vippagunta SR, Brittain HG, Grant DJ. Crystalline solids. Adv. Drug Deliv. Rev., 48, 3–26 (2001).
- 12) Thakuria R, Delori A, Jones W, Lipert MP, Roy L, Rodriguez-Hornedo N. Pharmaceutical cocrystals and poorly soluble drugs. Int. J. Pharm., 453, 101–125 (2013).
- 13) Lin Y, Yang H, Yang C, Wang J. Preparation, characterization, and evaluation of dipfluzine-benzoic acid co-crystals with improved physicochemical properties. Pharm. Res., 31, 566–578 (2014).
- 14) Fukami T. Current status and promising future of pharmaceutical cocrystals in development of oral dosage forms. Nippon Yakurigaku Zasshi, 150, 36–40 (2017).
- 15) Hickey MB, Peterson ML, Scoppettuolo LA, Morrisette SL, Vetter A, Guzman H, Remenar JF, Zhang Z, Tawa MD, Haley S, Zaworotko MJ, Almarsson O. Performance comparison of a co-crystal of carbamazepine with marketed product. Eur. J. Pharm. Biopharm., 67, 112–119 (2007).
- 16) Friščič T, Trask AV, Jones W, Motherwell WD. Screening for inclusion compounds and systematic construction of three-component solids by liquid-assisted grinding. Angew. Chem. Int. Ed. Engl., 45, 7546–7550 (2006).
- 17) Tumanov IA, Michalchuk AAL, Politov AA, Boldyreva EV, Boldyrev VV. Inadvertent liquid assisted grinding: a key to “dry” organic mechano-co-crystallisation? CrystEngComm, 19, 2830–2835 (2017).
- 18) Hasa D, Miniussi E, Jones W. Mechanochemical synthesis of multicomponent crystals: one liquid for one polymorph? A myth to dispel. Cryst. Growth Des., 16, 4582–4588 (2016).
- 19) Setyawan D, Sari R, Yusuf H, Primaharinastiti R. Preparation and characterization of artesunate-nicotinamide cocrystal by solvent evaporation and slurry method. Asian J. Pharm. Clin. Res., 7, 62–65 (2014).
- 20) Takata N, Shiraki K, Takano R, Hayashi Y, Terada K. Cocrystal screening of stanolone and mestanolone using slurry crystallization. Cryst. Growth Des., 8, 3032–3037 (2008).
- 21) Rodríguez-Hornedo N, Nehm SJ, Seefeldt KF, Pagan-Torres Y, Falkiewicz CJ. Reaction crystallization of pharmaceutical molecular complexes. Mol. Pharm., 3, 362–367 (2006).
- 22) Zhang GG, Henry RF, Borchardt TB, Lou X. Efficient co-crystal screening using solution-mediated phase transformation. J. Pharm. Sci., 96, 990–995 (2007).
- 23) Apshingekar PP, Aher S, Kelly AL, Brown EC, Paradkar A. Synthesis of caffeine/maleic acid co-crystal by ultrasound-assisted slurry co-crystallization. J. Pharm. Sci., 106, 66–70 (2017).
- 24) Bučar D-K, Henry RF, Lou X, Duerst RW, MacGillivray LR, Zhang GGZ. Cocrystals of caffeine and hydroxybenzoic acids composed of multiple supramolecular heterosynthons: screening via solution-mediated phase transformation and structural characterization. Cryst. Growth Des., 9, 1932–1943 (2009).
- 25) do Amaral LH, do Carmo FA, Amaro MI, de Sousa VP, da Silva L, de Almeida GS, Rodrigues CR, Healy AM, Cabral LM. Development and characterization of dapsone cocrystal prepared by scalable production methods. AAPS PharmSciTech, 19, 2687–2699 (2018).
- 26) Guo M, Wang K, Qiao N, Yardley V, Li M. Investigating permeation behavior of flufenamic acid cocrystals using a dissolution and permeation system. Mol. Pharm., 15, 4257–4272 (2018).
- 27) Khatioda R, Bora P, Sarma B. Trimorphic ethenzamide cocrystal: in vitro solubility and membrane efflux studies. Cryst. Growth Des., 18, 4637–4645 (2018).
- 28) Khatioda R, Saikia B, Das PJ, Sarma B. Solubility and in vitro drug permeation behavior of ethenzamide cocrystals regulated in physiological pH environments. CrystEngComm, 19, 6992–7000 (2017).
- 29) Bommaka MK, Mannava MC, Suresh K, Gunnam A, Nangia A. Entacapone: improving aqueous solubility, diffusion permeability, and cocrystal stability with theophylline. Cryst. Growth Des., 18, 6061–6069 (2018).
- 30) Surov AO, Volkova TV, Churakov AV, Proshin AN, Terekhova IV, Perlovich GL. Cocrystal formation, crystal structure, solubility and permeability studies for novel 1,2,4-thiadiazole derivative as a potent neuroprotector. Eur. J. Pharm. Sci., 109, 31–39 (2017).
- 31) Mochalin V, Osswald S, Gogotsi Y. Contribution of functional groups to the Raman spectrum of nanodiamond powders. Chem. Mater., 21, 273–279 (2009).
- 32) Otaki T, Tanabe Y, Kojima T, Miura M, Ikeda Y, Koide T, Fukami T. In situ monitoring of cocrystals in formulation development using low-frequency Raman spectroscopy. Int. J. Pharm., 542, 56–65 (2018).
- 33) Childs SL, Stahly GP, Park A. The salt–cocrystal continuum: the influence of crystal structure on ionization state. Mol. Pharm., 4, 323–338 (2007).
- 34) Press B. Optimization of the Caco-2 permeability assay to screen drug compounds for intestinal absorption and efflux. Permeability Barrier: Methods and Protocols. (Turksen K ed.) Humana Press, Totowa, NJ, pp. 139–154 (2011).
- 35) Angelis ID, Turco L. Caco-2 cells as a model for intestinal absorption. Curr Protoc Toxicol, 47, 20.6.1–20.6.15 (2011).
- 36) Failla ML, Chitchumronchokchai C, Ferruzzi MG, Goltz SR, Campbell WW. Unsaturated fatty acids promote bioaccessibility and basolateral secretion of carotenoids and α-tocopherol by Caco-2 cells. Food Funct, 5, 1101–1112 (2014).
- 37) Yang C, Fischer M, Kirby C, Liu K, Zhu H, Zhang H, Chen Y, Sun Y, Zhang L, Tsao R. Bioaccessibility, cellular uptake and transport of luteins and assessment of their antioxidant activities. Food Chem., 249, 66–76 (2018).
- 38) Gujral N, Suh JW, Sunwoo HH. Effect of anti-gliadin IgY antibody on epithelial intestinal integrity and inflammatory response induced by gliadin. BMC Immunol., 16, 41 (2015).
- 39) Ilboudo S, Fouche E, Rizzati V, Toe AM, Gamet-Payrastre L, Guissou PI. In vitro impact of five pesticides alone or in combination on human intestinal cell line Caco-2. Toxicol. Rep, 1, 474–489 (2014).
- 40) Xiao J, Högger P. Stability of dietary polyphenols under the cell culture conditions: avoiding erroneous conclusions. J. Agric. Food Chem., 63, 1547–1557 (2015).
- 41) Kaldas MI, Walle UK, Walle T. Resveratrol transport and metabolism by human intestinal Caco-2 cells. J. Pharm. Pharmacol., 55, 307–312 (2003).
- 42) Yan Y, Chen J-M, Lu T-B. Simultaneously enhancing the solubility and permeability of acyclovir by crystal engineering approach. CrystEngComm, 15, 6457–6460 (2013).