Abstract
Transporter gene knockout models are a practical and widely used tool for pharmacokinetic studies in drug discovery. P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp) are major efflux transporters that control absorption and bioavailability, and are important when determining oral drug disposition. To the best of our knowledge, beyond the rule of five (bRo5) molecules launched on the market to date tend to be substrates for efflux transporters. The purpose of this study is to evaluate in vivo the impact of efflux transporters on the oral absorption process and systemic clearance using rats which lack P-gp and/or Bcrp expression. We administered five bRo5 substrates (asunaprevir, cyclosporine, danoprevir, ledipasvir, and simeprevir) intravenously or orally to wild-type and Mdr1a, Bcrp, and Mdr1a/Bcrp knockout rats, calculated the clearance, oral bioavailability, and absorption rate profile of each substrate, and compared the results. Systemic clearance of the substrates in knockout rats changed within approximately ±40% compared to wild-types, suggesting the efflux transporters do not have a significant influence on clearance in rats. On the other hand, the oral absorption of substrates in the knockout rats, especially those lacking Mdr1a, increased greatly—between 2- and 5-fold more than in wild-types. This suggests that rat efflux transporters, especially P-gp, greatly reduce the oral exposure of these substrates. Moreover, results on the absorption rate–time profile suggest that efflux transporters are constantly active during the absorption period in rats. Transporter knockout rats are a useful in vivo tool for estimating the transporter-mediated disposition of bRo5 molecules in drug discovery.
INTRODUCTION
Since it was first formulated in 1997, Lipinski's rule of five (Ro5)1) has been widely proposed as a qualitative predictive model for oral absorption trends. The Ro5 outlined that 90% of oral drugs in use at that time obeyed three out of four of the following guidelines: molecular weight (MW) ≤ 500 Da, c Log P ≤5, H-bond donors (HBD) ≤ 5, and H-bond acceptors (HBA) ≤ 10.1,2) By contrast, in drug discovery today, difficult targets with large and flat binding sites are often better suited to molecules beyond the rule of 5 (bRo5).2) Consequently, the trend of drug discovery in chemistry has shifted from Ro5 molecules to bRo5 molecules such as cyclic peptides. However, increased MW is known to be correlated with decreased solubility, decreased permeability, and increased transporter-mediated efflux.2) Thus, bRo5 molecules tend to have poor pharmacokinetics, such as low bioavailability.
ATP-binding cassette (ABC) transporters are membrane-embedded proteins that mediate the uptake or export of an enormous variety of substrates via an active energy-dependent mechanism. Drug efflux transporters of the ABC family limit the intracellular concentration of substrate agents by pumping them out of cells. P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp) are important drug efflux transporters located in the apical domain of the enterocyte of the lower gastro-intestinal tract, thereby limiting the absorption of drug substrates from the gastro-intestinal tract.3)
Transporter gene knockout models can provide useful information for understanding transporter-limited or transporter-mediated drug absorption, distribution, and excretion.4) Pharmacokinetic data obtained from studies using transporter knockout models are helpful for determining the impact of transporters in both pre-clinical compound selection and clinical compound-selection. So far, there have been many studies on transporter-mediated disposition using transporter gene knockout mice and rats.5) Transporter gene knockout rats, in particular, are a useful tool for pharmacokinetic studies for several reasons: first, rats have sufficient blood volume and other relevant bodily fluids for ease of surgical operation. Secondly, rats can be used to predict oral drug absorption in the human small intestine because of their similar drug intestinal absorption profiles and transporter expression patterns.6) Thirdly, there have been several reports on SAGE Mdr1a and Bcrp knockout rats that demonstrate modest compensatory changes in gene expression and overall pathology7) as well as the absorption, distribution, metabolism, and excretion (ADME) of various transporter substrates.8–11) While P-gp knockout mice may be more widely used to evaluate pharmacokinetics, their compensatory changes in gene expression and pathology have not been reported.
To the best of our knowledge, many bRo5 molecules (MW >700 Da) launched on the market to date are substrates for efflux transporters. The purpose of this study is to evaluate in vivo the impact of efflux transporters on the oral absorption process and systemic clearance of bRo5 molecules using rats which lack P-gp and/or Bcrp expression.
MATERIALS AND METHODS
MaterialsAsunaprevir and danoprevir were purchased from ChemScene (Monmouth Junction, NJ, U.S.A.). Cyclosporine was purchased from Sigma (St. Louis, MO, U.S.A.). Simeprevir was purchased from MedChemExpress (Monmouth, NJ, U.S.A.). Ledipasvir was purchased from Selleck Chemicals LLC (Houston, TX, U.S.A.). All other chemicals were of reagent grade and were readily available from commercial sources.
AnimalsMale Sprague–Dawley wild-type and Mdr1a, Bcrp, and Mdr1a/Bcrp knockout rats purchased from SAGE Labs, Sigma Life Science (St. Louis, MO, U.S.A.). The rats, aged from 7 to 14 weeks, were used in this study. The animals had free access to food and water, and were housed in an environmentally controlled room with temperature maintained at 23 ± 3°C, humidity at 50 ± 20%, and a 12-h light/dark cycle (lights on 07 : 00–19 : 00). Care of the animals and the protocol used complied with the “General Considerations for Animal Experiments” promulgated in Chugai Pharmaceutical Co., Ltd. and were approved by the Institutional Animal Care and Use Committee, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
Pharmacokinetic StudiesWe administered five substrates (asunaprevir, cyclosporine, danoprevir, ledipasvir, and simeprevir) intravenously or orally to wild-type and Mdr1a, Bcrp, and Mdr1a/Bcrp knockout rats. Intravenous and oral doses of each substrate in the pharmacokinetic study are shown in Table 1.
Table 1. Outline of the Pharmacokinetic Study
| Substrate | Route | Dose (mg/kg) | Volume (mL/kg) | Vehicle |
|---|
| Asunaprevir | Oral | 2 | 2 | 10% (v/v) DMSO and 10% (v/v) Cremophor EL in distilled water |
| Intravenous | 1 | 1 | 10% (v/v) DMSO and 10% (v/v) Cremophor EL in saline |
| Cyclosporine | Oral | 1 | 10 | 10% (v/v) DMSO and 10% (v/v) Cremophor EL in distilled water |
| Intravenous | 1 | 2 | 10% (v/v) DMSO and 10% (v/v) Cremophor EL in saline |
| Danoprevir | Oral | 5 | 5 | 10% (v/v) DMSO and 10% (v/v) Cremophor EL in distilled water |
| Intravenous | 1 | 1 | 10% (v/v) DMSO and 10% (v/v) Cremophor EL in saline |
| Ledipasvir | Oral | 2 | 2 | 10% (v/v) DMSO and 10% (v/v) Cremophor EL in distilled water |
| Intravenous | 1 | 1 | 10% (v/v) DMSO and 10% (v/v) Cremophor EL in saline |
| Simeprevir | Oral | 2 | 2 | 10% (v/v) DMSO and 10% (v/v) Cremophor EL in distilled water |
| Intravenous | 1 | 1 | 10% (v/v) DMSO and 10% (v/v) Cremophor EL in saline |
Blood samples were collected at the following times after dose administration: 0.033 (intravenous only), 0.167, 0.5, 1, 2, 4, 8, and 24 h, except for cyclosporine, which was collected at 0.05 (intravenous only), 0.25 (oral only), 0.5, 1, 2, 4, 7, 24, and 48 h. Plasma was prepared by centrifuging the blood at 4°C at 13000 × g for 5 min and freezing at −30°C until analysis.
BioanalysisPlasma samples were analyzed by LC-MS/MS after protein precipitation with acetonitrile. The LC-MS/MS conditions were as follows: the instrument was an Acquity ultra performance liquid chromatography (UPLC) system (Waters Corp., Milford, MA, U.S.A.) equipped with a 4000QTRAP system (AB SCIEX, Framingham, MA, U.S.A.); the column was an ACQUITY UPLCBEH C18 (2.1 × 30 mm, 1.7 µm) at a column temperature of 40–60°C; the mobile phase was operated under gradient conditions and consisted of 0.1% (v/v) formic acid in water (A) and 0.1% (v/v) formic acid in acetonitrile. Mass spectrometry detection was performed by positive ionization electrospray. The selective reaction monitoring mode was used to monitor ions as follows (m/z: precursor ion/production): asunaprevir (748.30/535.10), cyclosporine (1203.00/156.20), danoprevir (732.30/68.10), ledipasvir (889.70/732.45), simeprevir (750.60/315.00), and an internal standard indomethacin (358.37/139.14).
Pharmacokinetic AnalysisPharmacokinetic parameters were calculated by noncompartmental analysis using Phoenix WinNonlin version 8.0 software (Pharsight Corp., Mountain View, CA, U.S.A.). Bioavailability was calculated by comparing the dose-normalized area under the curve (AUC) after intravenous and oral administration. Absorption rate–time profiles were calculated using the deconvolution method (Excel 2013, Microsoft, Redmond, WA, U.S.A.). All statistical analyses were performed using one-way ANOVA with the Dunnett multiple comparisons post-hoc test (GraphPad Prism version 7.04 software, GraphPad Software, Inc., San Diego, CA, U.S.A.)
RESULTS
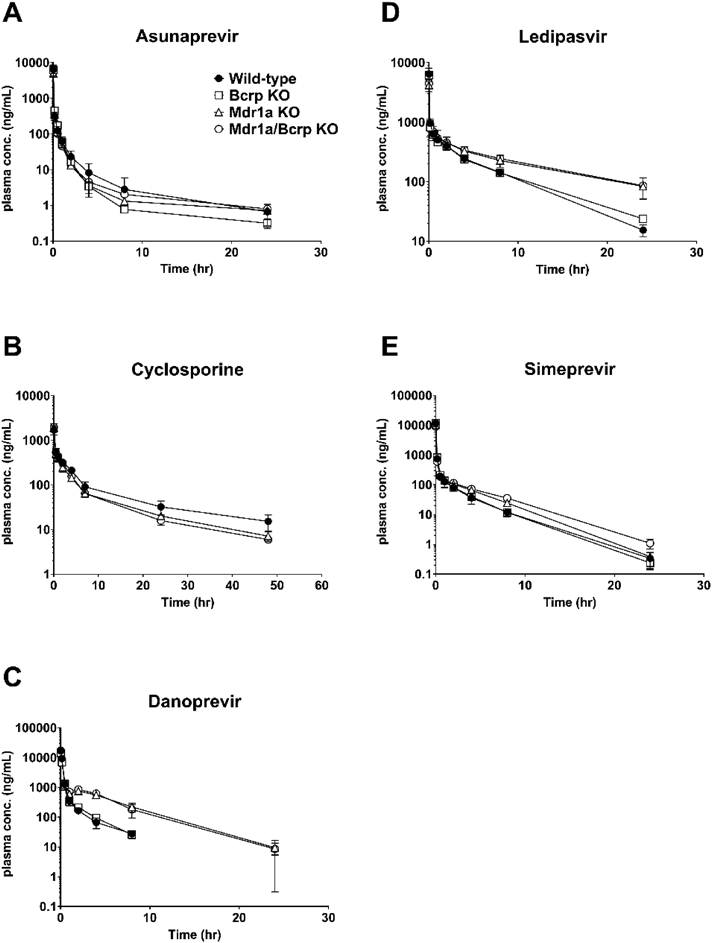
The pharmacokinetics of asunaprevir are summarized in Figs. 1A, 2A, and Table 2. In Mdr1a, Bcrp, and Mdr1a/Bcrp knockout rats, there was almost no difference in systemic clearance, distribution volume, and half-life compared with wild-type rats. Oral AUC and bioavailability showed a respectively 2.1-fold and 2.5-fold greater increase in Mdr1a knockout rats, and a respectively 3.1-fold and 3.8-fold greater increase in Mdr1a/Bcrp knockout rats. Asunaprevir Cmax increased 2.0-fold in Mdr1a/Bcrp knockout rats, and the Tmax in all the knockout rats was almost the same as in wild-types. In Bcrp knockout rats, asunaprevir pharmacokinetics were almost the same compared with wild-types.
Table 2. Pharmacokinetic Parameters of Five Substrates in Wild-Type, Bcrp Knockout, Mdr1a Knockout, and Mdr1a/Bcrp Knockout Rats
| Parameter | Wild-type | Bcrp KO | Mdr1a KO | Mdr1a/Bcrp KO |
|---|
| Asunaprevir (1 mg/kg, i.v.) | | | | |
| AUCinf (ng·h/mL) | 821 ± 239 | 800 ± 60 | 678 ± 74 | 683 ± 107 |
| CL (mL/min/kg) | 21.4 ± 5.3 | 20.9 ± 1.7 | 24.8 ± 3 | 24.8 ± 3.7 |
| Vss (L/kg) | 1.75 ± 0.66 | 0.936 ± 0.291 | 4.43 ± 4.79 | 2.29 ± 0.79 |
| T1/2 (h) | 11.8 ± 5.7 | 11.8 ± 4 | 20.1 ± 16.2 | 10.5 ± 0.8 |
| Asunaprevir (2 mg/kg, p.o.) | | | | |
| AUCinf (ng·h/mL) | 38.2 ± 7.7 | 39.5 ± 8 | 79.2 ± 18.4** | 120 ± 22** |
| Cmax (ng/mL) | 26.2 ± 5.2 | 24.3 ± 6.8 | 24.8 ± 8.8 | 52.1 ± 14.6** |
| Tmax (h) | 0.417 ± 0.167 | 0.334 ± 0.192 | 0.875 ± 0.75 | 0.5 ± 0 |
| T1/2 (h) | 7.12 ± 1.4 | 7.93 ± 1.88 | 8.07 ± 1.5 | 13.5 ± 4.4* |
| F (%) | 2.33 ± 0.47 | 2.47 ± 0.5 | 5.85 ± 1.36** | 8.79 ± 1.59** |
| Cyclosporine (1 mg/kg, i.v.) | | | | |
| AUCinf (ng·h/mL) | 3960 ± 760 | | 2880 ± 470* | 2900 ± 260* |
| CL (mL/min/kg) | 4.33 ± 0.86 | | 5.92 ± 1.07* | 5.79 ± 0.53 |
| Vss (L/kg) | 3.96 ± 0.92 | | 3.92 ± 0.76 | 3.23 ± 0.47 |
| T1/2 (h) | 16.1 ± 2.5 | | 13.1 ± 1.4 | 12.2 ± 0.3* |
| Cyclosporine (1 mg/kg, p.o.) | | | | |
| AUCinf (ng·h/mL) | 546 ± 250 | | 210 ± 140** | 1260 ± 320** |
| Cmax (ng/mL) | 73.9 ± 30.8 | | 122 ± 22 | 149 ± 34* |
| Tmax (h) | 2 ± 0 | | 2.5 ± 1 | 2.5 ± 1 |
| T1/2 (h) | 5.8 ± 1.25 | | 9.12 ± 0.86** | 8.52 ± 1.45* |
| F (%) | 13.8 ± 6.3 | | 41.9 ± 5** | 43.3 ± 11.2** |
| Danoprevir (1 mg/kg, i.v.) | | | | |
| AUCinf (ng·h/mL) | 4710 ± 240 | 4170 ± 510 | 8330 ± 1000** | 8330 ± 1350** |
| CL (mL/min/kg) | 3.54 ± 0.18 | 4.04 ± 0.47 | 2.02 ± 0.24** | 2.04 ± 0.29** |
| Vss (L/kg) | 0.185 ± 0.033 | 0.234 ± 0.046 | 0.413 ± 0.103** | 0.369 ± 0.056* |
| T1/2 (h) | 2.91 ± 0.29 | 2.13 ± 0.26* | 3.51 ± 0.31 | 3.44 ± 0.48 |
| Danoprevir (5 mg/kg, p.o.) | | | | |
| AUCinf (ng·h/mL) | 9340 ± 2760 | 11000 ± 2900 | 36700 ± 5200** | 43800 ± 4200** |
| Cmax (ng/mL) | 6880 ± 290 | 7310 ± 680 | 14000 ± 4200* | 15100 ± 2400** |
| Tmax (h) | 0.667 ± 0.289 | 0.5 ± 0 | 0.75 ± 0.289 | 0.5 ± 0 |
| T1/2 (h) | 1.37 ± 0.19 | 2.84 ± 0.94* | 2.87 ± 0.28** | 3.08 ± 0.24** |
| F (%) | 39.7 ± 11.7 | 52.6 ± 13.7 | 88 ± 12.5** | 105 ± 10** |
| Ledipasvir (1 mg/kg, i.v.) | | | | |
| AUCinf (ng·h/mL) | 4060 ± 440 | 4170 ± 300 | 7000 ± 2040** | 7180 ± 720** |
| CL (mL/min/kg) | 4.14 ± 0.42 | 4.01 ± 0.3 | 2.53 ± 0.67** | 2.34 ± 0.25** |
| Vss (L/kg) | 1.39 ± 0.13 | 1.64 ± 0.13 | 1.99 ± 0.41* | 1.87 ± 0.19* |
| T1/2 (h) | 4.99 ± 0.3 | 6.18 ± 0.28 | 10.6 ± 2.1** | 10.4 ± 0.5** |
| Ledipasvir (2 mg/kg, p.o.) | | | | |
| AUCinf (ng·h/mL) | 505 ± 160 | 685 ± 71 | 2870 ± 430** | 2380 ± 410** |
| Cmax (ng/mL) | 39.2 ± 12.3 | 49.3 ± 2.1 | 112 ± 4** | 110 ± 42** |
| Tmax (h) | 4 ± 0 | 4 ± 0 | 6 ± 2.31 | 6 ± 2.31 |
| T1/2 (h) | 7.04 ± 0.88 | 7.89 ± 0.55 | 14.5 ± 2.8** | 15.6 ± 4.3** |
| F (%) | 6.23 ± 1.97 | 8.21 ± 0.85 | 20.5 ± 3.1** | 16.6 ± 2.8** |
| Simeprevir (1 mg/kg, i.v.) | | | | |
| AUCinf (ng·h/mL) | 1650 ± 290 | 1690 ± 170 | 1770 ± 80 | 1710 ± 260 |
| CL (mL/min/kg) | 10.4 ± 1.9 | 9.92 ± 0.98 | 9.42 ± 0.41 | 9.93 ± 1.74 |
| Vss (L/kg) | 0.656 ± 0.113 | 0.617 ± 0.07 | 0.916 ± 0.129 | 1.41 ± 0.29** |
| T1/2 (h) | 3.07 ± 0.46 | 2.8 ± 0.2 | 2.68 ± 0.09 | 3.21 ± 0.15 |
| Simeprevir (2 mg/kg, p.o.) | | | | |
| AUCinf (ng·h/mL) | 351 ± 163 | 426 ± 169 | 1710 ± 600** | 1790 ± 340** |
| Cmax (ng/mL) | 167 ± 102 | 174 ± 44 | 357 ± 144* | 342 ± 50 |
| Tmax (h) | 1 ± 0 | 1 ± 0 | 1.75 ± 0.5* | 1.75 ± 0.5* |
| T1/2 (h) | 5.44 ± 4.67 | 2.15 ± 1.3 | 2.51 ± 0.08 | 3.11 ± 0.31 |
| F (%) | 10.6 ± 5 | 12.6 ± 5 | 48.2 ± 16.8** | 52.2 ± 9.8** |
KO, knockout; AUCinf, area under the curve extrapolated to infinity; CL, clearance; Vss, volume of distribution; T1/2, half-life; Cmax, maximum concentration; Tmax, time of maximum concentration; F, bioavailability. * p < 0.05, knockout versus wild-type rats. ** p < 0.01, knockout versus wild-type rats.
The pharmacokinetics of cyclosporine are summarized in Figs. 1B, 2B, and Table 2. Cyclosporine systemic clearance increased approximately 35% in Mdr1a and Mdr1a/Bcrp knockout rats. Cyclosporine half-life was slightly shorter in Mdr1a/Bcrp knockout rats, and the distribution volume in almost all knockout rats was the same as in wild-types. Oral AUC and bioavailability showed a respectively 2.2- and 3.0-fold greater increase in Mdr1a knockout rats, and a respectively 2.3- and 3.1-fold greater increase in Mdr1a/Bcrp knockout rats. Cyclosporine Cmax was increased 2.0-fold in Mdr1a/Bcrp knockout rats, and the Tmax in all knockout rats was almost the same as in wild-types. In Bcrp knockout rats, cyclosporine pharmacokinetics were almost the same compared with wild-types.
The pharmacokinetics of danoprevir are summarized in Figs. 1C, 2C, and Table 2. In Mdr1a and Mdr1a/Bcrp knockout rats, danoprevir systemic clearance was approximately 40% reduced, and the distribution volume was increased about 2-fold. It is possible that enterohepatic recycling occurs by biliary excretion and intestinal reabsorption in Mdr1a and Mdr1a/Bcrp knockout rats; this is supported by rat excretion studies, suggesting that hepatic elimination via biliary excretion is the major clearance pathway for danoprevir.12) Danoprevir half-life was mostly comparable to wild-types. Oral AUC, bioavailability, and Cmax respectively increased 3.9-, 2.2-, and 2.0-fold in Mdr1a knockout rats, and 4.7-, 2.6-, and 2.2-fold in Mdr1a/Bcrp knockout rats. Danoprevir Tmax in all knockout rats was almost the same as in wild-types. In Bcrp knockout rats, danoprevir pharmacokinetics were almost the same compared with wild-types.
The pharmacokinetics of ledipasvir are summarized in Figs. 1D, 2D, and Table 2. In Mdr1a and Mdr1a/Bcrp knockout rats, ledipasvir systemic clearance was about 40% reduced, and the distribution volume was increased 1.3- to 1.4-fold, resulting in a 2-fold longer half-life. The reduced clearance may be because ledipasvir excretion in bile as an unchanged parent drug was a major route of elimination.13) Oral AUC, bioavailability, and Cmax respectively increased 5.7-, 3.3-, and 2.9-fold in Mdr1a knockout rats, and 4.7-, 2.7-, and 2.8-fold in Mdr1a/Bcrp knockout rats. The Tmax in all knockout rats was almost the same as in wild-types. In Bcrp knockout rats, ledipasivir pharmacokinetics were almost the same compared with wild-types.
The pharmacokinetics of simeprevir are summarized in Figs. 1E, 2E, and Table 2. In Mdr1a, Bcrp and Mdr1a/Bcrp knockout rats, simeprevir systemic clearance was almost the same as in wild-types. Simeprevir distribution volume increased 2.1-fold in Mdr1a/Bcrp knockout rats. Oral AUC, bioavailability and Cmax respectively increased 4.9-, 4.5-, and 2.1-fold in Mdr1a knockout rats, and 5.1-, 4.9-, and 2.0-fold in Mdr1a/Bcrp knockout rats. The Tmax was slightly longer in Mdr1a and Mdr1a/Bcrp knockout rats. In Bcrp knockout rats, simeprevir pharmacokinetics were almost the same compared with wild-types.
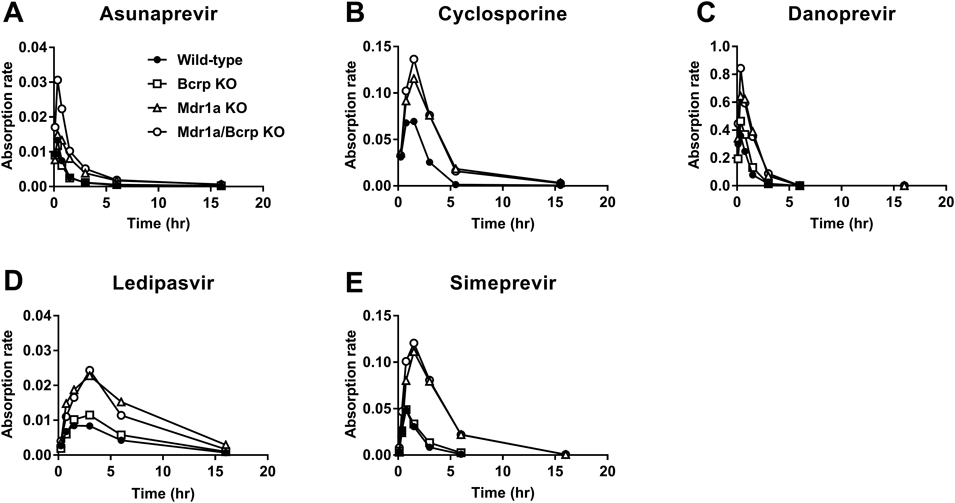
Absorption rate-time profiles of asunaprevir, cyclosporine, danoprevir, ledipasvir, and simeprevir are summarized in Fig. 3. The maximum absorption rate of each substrate is shown as follows (wild-type/Bcrp KO/Mdr1a KO/Mdr1a-Bcrp KO or wild-type/Mdr1a KO/Mdr1a-Bcrp KO (cyclosporine only)): asunaprevir (0.013/0.012/0.015/0.031), cyclosporine (0.069/0.12/0.14), danoprevir (0.36/0.46/0.64/0.84), ledipasvir (0.0084/0.011/0.023/0.024), and simeprevir (0.048/0.049/0.11/0.12). Absorption time varied depending on the substrate, but not significantly on the kind of rat. The absorption rate-time profile in the Mdr1a and/or Mdr1a/Bcrp knockout rats was higher than in wild-types at almost all time points. These results suggest that efflux transporters are constantly active during the absorption time in rats.
DISCUSSION
The aim of this study is to evaluate in vivo the impact of efflux transporters on the oral absorption process and systemic clearance of five bRo5 substrates using wild-type and Mdr1a, Bcrp, and Mdr1a/Bcrp knockout rats. P-gp and Bcrp are major efflux transporters that control absorption and bioavailability, and are important when determining oral drug disposition. In particular, P-gp is important in drug-like ADME mostly because of the striking diversity of substrate structures that it is able to transport, including a vast number of drugs with a range of therapeutic applications.14) In the present study we used five bRo5 molecules: asunaprevir, cyclosporine, and danoprevir are P-gp substrates, and ledipasvir and simeprevir are both P-gp and Bcrp substrates. Their ADME-related properties are as follows: asunaprevir is a substrate for CYP3A, P-gp, organic anion transporting polypeptide (OATP) 1B1, and OATP2B1.15) Cyclosporine is a substrate for CYP3A and P-gp.16) Danoprevir is a substrate for CYP3A, P-gp, OATP1B1, OATP1B3, and multidrug resistance associated proteins (MRP2).17,18) Ledipasvir is a substrate for P-gp and BCRP.19) Simeprevir is a substrate for CYP3A, P-gp, BCRP, OATP1B1, OATP1B3, and MRP2.20)
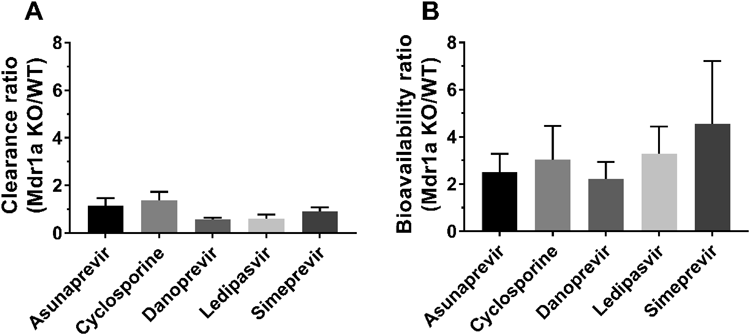
Systemic clearance of the substrates in Mdr1a knockout rats changed within approximately ±40% (range, +43% to −37%) compared to wild-types. These results indicate that P-gp does not have a significant influence on systemic clearance in rats when enterohepatic recirculation is taken into account. P-gp is found not only on the apical surface of columnar epithelial cells of the small and large intestine, but also on the biliary canalicular membrane of hepatocytes and the apical surface of epithelial cells of the proximal tubules of kidneys, as well as on the apical surface of epithelial cells of the placenta and the apical surface of endothelial cells in blood capillaries in the brain.21,22) Therefore P-gp can enhance the elimination of drugs from hepatocytes, renal tubules, and intestinal epithelial cells into the adjacent luminal space.23) The decreased systemic clearance of danoprevir observed in Mdr1a knockout rats may be caused by both the suppression of biliary excretion and reabsorption from the intestine, considering its main elimination pathway and its second peak in plasma concentration-time profiles after intravenous administration (Fig. 1C). The decreased systemic clearance of ledipasvir observed in Mdr1a knockout rats may be caused by the suppression of biliary excretion and direct intestinal secretion, considering its main elimination pathway. The increased systemic clearance of cyclosporine observed in Mdr1a knockout rats was unexpected because our unpublished data demonstrated no change in cyclosporine clearance with a concomitant oral dose of zosuquidar (a P-gp specific inhibitor) in rats. This increased clearance may be influenced by compensatory changes in SAGE Mdr1a knockout rats.7) On the other hand, systemic clearance of the substrates in the Bcrp knockout rats hardly changed compared to wild-types.
The oral bioavailability of the substrates in the knockout rats, especially Mdr1a knockout rats, increased greatly—between 2.2- and 4.5-fold more than in wild-types. These results indicate that P-gp has a significant influence on oral absorption in rats. Moreover, the absorption rate-time profile in the Mdr1a knockout rats was higher than in wild-types at almost all time points. These results suggest that P-gp is constantly active during absorption in rats. A previous report suggested that P-gp plays a minimal role in the in vivo intestinal absorption process of verapamil due to high water solubility and high membrane permeability.24) On the other hand, transport of drugs with moderate passive permeability is highly attenuated by P-gp.25) The five bRo5 substrates can be classified using the BCS system (Biopharmaceutics Classification System), which is a framework for broadly classifying oral drugs into four categories based on their aqueous solubility and permeability across biological membranes.26) Asunaprevir and ledipasvir are classified as BCS II (low solubility/high permeability) drugs. Cyclosporine, danoprevir, and simeprevir are classified as BCS IV (low solubility/low permeability) drugs. Therefore our pharmacokinetic results in Mdr1a knockout rats using bRo5 substrates demonstrate that P-gp limits the in vivo intestinal absorption of high to low permeability P-gp substrates. The fact that the change in oral bioavailability was larger than in systemic clearance suggests that P-gp has a greater impact on absorption than on excretion (Fig. 4). A synergistic effect between P-gp and Bcrp on intestinal absorption was not observed in this study.
To investigate the extrapolation of this efflux transporter-mediated disposition to humans, we collected information from clinical studies related to P-gp and Bcrp mediated drug–drug interactions (DDIs) using a metabolism and transport drug interaction database (University of Washington, Seattle, WA, U.S.A.). We listed clinical studies on each of the five substrates which demonstrate the maximum P-gp-mediated DDI (because of the comparison with knockout rats) in Table 3. The clinical percent change in the AUC of each substrate does not correlate quantitatively to the increased oral absorption in the P-gp knockout rats. There are two main reasons for this: first, the lack of information from clinical settings makes it difficult to investigate the in vivo correlation between humans and experimental animals. In fact, ketoconazole and telaprevir27) are inhibitors not only of P-gp, but also CYP3A, suggesting the clinical percent change in the AUC of both asunaprevir and cyclosporine may include CYP3A-mediated DDI. Similarly, ritonavir is an inhibitor of CYP3A, OATP1B1, OATP1B3, and P-gp, suggesting the clinical percent change in the AUC of danoprevir may include CYP3A-mediated DDI and hepatic first-pass effects.18) As stated above, there are limitations in obtaining pure P-gp-mediated DDI information in a clinical setting. Secondly, species differences in the expression, substrate specificity, and inhibition of P-gp have been reported. P-gp mRNA expression is more enriched in the gastrointestinal tract of rodents compared with humans.28) The apparent Km value for diltiazem exhibited an approximately 16.5-fold difference among seven cell lines including humans and rats.29) Correlation of the efflux ratios between MDR1 stably expressing cells from various animal species was also reported, showing that the correlation coefficient between human MDR1 and rat MDR1a is 0.762.30) In addition, P-gp humanized mice have recently become available and are able to elucidate species differences for P-gp between humans and mice in vivo.31) On the other hand, there is no clinical study on Bcrp-mediated DDI using ledipasvir and simeprevir in the database, suggesting that the two substrates do not have Bcrp-mediated DDI risks. These facts are consistent with our results showing that oral absorption of the substrates in Bcrp knockout rats increases slightly, just 1.4-fold (ledipasvir) and 1.2-fold (simeprevir) more than in wild-types. It may be possible to evaluate human P-gp and/or Bcrp mediated DDI risk using P-gp and/or Bcrp knockout rats, but not to quantitatively evaluate human P-gp and/or Bcrp mediated absorption. Although transporter knockout animals are powerful tools for studying drug absorption and disposition, there are limitations to their use in quantitatively predicting human pharmacokinetics.
Table 3. Clinical Information Related to P-gp Inhibition
| Substrate | Route (substrate) | Inhibitor | % Change in AUC | Reference |
|---|
| Asunaprevir | Oral | Ketoconazole | 860 | 33) |
| Cyclosporine | Oral | Telaprevir | 354 | 34) |
| Danoprevir | Oral | Ritonavir | 227 | 18) |
| Ledipasvir | Oral | Verapamil | 66 | Label information of Harvoni (NDA #205834) |
| Simeprevir | Oral | Ledipasvir | 184 | Label information of Harvoni (NDA #205834) |
The clinical information was collected from DIDB (The University of Washington Metabolism and Transport Drug Interaction Database, copyright University of Washington 2005–2019). Accessed: 30 June 2019.
As the trend of drug discovery in chemistry has shifted from Ro5 molecules to bRo5 molecules, we have been facing more drug-likeness issues including the reliability of in vitro evaluations and human predictability. Regarding ADME, bRo5 molecules tend to show poor solubility, poor permeability, and susceptibility to transporter-mediated efflux. In fact, several clinical studies of cyclosporine and paclitaxel clearly indicate that P-gp does play a significant role in limiting their oral absorption, implying that the poor water solubility and slow dissolution rate can result in low drug concentration in the intestinal lumen relative to their Km value for P-gp transport, and that the large molecular size can impede the rate of passive diffusion across cell membranes.32) Therefore, in order to appropriately select clinical candidates, we have to carefully estimate the efflux transporter-mediated absorption of bRo5 molecules while taking into consideration their solubility, dissolution rate, and passive diffusion. In this situation, we evaluated in vivo the impact of efflux transporters on the oral absorption process and systemic clearance of bRo5 molecules using efflux transporter knockout rats.
In conclusion, we revealed that efflux transporters, especially P-gp, greatly reduce the oral exposure of five examined bRo5 substrates, and that efflux transporters are constantly active during absorption in rats. Transporter knockout rats are a useful in vivo tool for estimating the transporter-mediated disposition of bRo5 molecules in drug discovery.
Acknowledgments
The author would like to thank Motohiro Kato (formerly of Chugai Pharmaceutical Co., Ltd.) for his helpful suggestions covering the whole field of pharmacokinetics. The author also would like to thank Yuji Sakurai (Chugai Pharmaceutical Co., Ltd.) and Hidekazu Kitamura (Chugai Research Institute for Medical Science, Inc.) for performing the pharmacokinetic studies. The author would like to acknowledge the useful advice of Jacob Davis in the preparation of this article.
Conflict of Interest
The author declares no conflict of interest.
REFERENCES
- 1) Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev., 46, 3–26 (2001).
- 2) Matsson P, Doak BC, Over B, Kihlberg J. Cell permeability beyond the rule of 5. Adv. Drug Deliv. Rev., 101, 42–61 (2016).
- 3) Kim RB. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab. Rev., 34, 47–54 (2002).
- 4) Giacomini KM, Huang SM, Tweedie DJ, et al. Membrane transporters in drug development. Nat. Rev. Drug Discov., 9, 215–236 (2010).
- 5) Zamek-Gliszczynski MJ, Lee CA, Poirier A, Bentz J, Chu X, Ellens H, Ishikawa T, Jamei M, Kalvass JC, Nagar S, Pang KS, Korzekwa K, Swaan PW, Taub ME, Zhao P, Galetin A. ITC recommendations for transporter kinetic parameter estimation and translational modeling of transport-mediated PK and DDIs in humans. Clin. Pharmacol. Ther., 94, 64–79 (2013).
- 6) Cao X, Gibbs ST, Fang L, Miller HA, Landowski CP, Shin HC, Lennernas H, Zhong Y, Amidon GL, Yu LX, Sun D. Why is it challenging to predict intestinal drug absorption and oral bioavailability in human using rat model. Pharm. Res., 23, 1675–1686 (2006).
- 7) Zamek-Gliszczynski MJ, Goldstein KM, Paulman A, Baker TK, Ryan TP. Minor compensatory changes in SAGE Mdr1a (P-gp), Bcrp, and Mrp2 knockout rats do not detract from their utility in the study of transporter-mediated pharmacokinetics. Drug Metab. Dispos., 41, 1174–1178 (2013).
- 8) Zamek-Gliszczynski MJ, Bedwell DW, Bao JQ, Higgins JW. Characterization of SAGE Mdr1a (P-gp), Bcrp, and Mrp2 knockout rats using loperamide, paclitaxel, sulfasalazine, and carboxydichlorofluorescein pharmacokinetics. Drug Metab. Dispos., 40, 1825–1833 (2012).
- 9) Suzuki M, Komura H, Yoshikawa T, Enya S, Nagao A, Takubo H, Kogayu M. Characterization of gastrointestinal absorption of digoxin involving influx and efflux transporter in rats: application of mdr1a knockout (−/−) rats into absorption study of multiple transporter substrate. Xenobiotica, 44, 1039–1045 (2014).
- 10) Fuchs H, Kishimoto W, Gansser D, Tanswell P, Ishiguro N. Brain penetration of WEB 2086 (Apafant) and dantrolene in Mdr1a (P-glycoprotein) and Bcrp knockout rats. Drug Metab. Dispos., 42, 1761–1765 (2014).
- 11) Bundgaard C, Jensen CJ, Garmer M. Species comparison of in vivo P-glycoprotein-mediated brain efflux using mdr1a-deficient rats and mice. Drug Metab. Dispos., 40, 461–466 (2012).
- 12) Brennan BJ, Moreira SA, Morcos PN, Navarro MT, Asthappan J, Goelzer P, Weigl P, Smith PF. Pharmacokinetics of a three-way drug interaction between danoprevir, ritonavir and the organic anion transporting polypeptide (OATP) inhibitor ciclosporin. Clin. Pharmacokinet., 52, 805–813 (2013).
- 13) German P, Mathias A, Brainard D, Kearney BP. Clinical pharmacokinetics and pharmacodynamics of Ledipasvir/Sofosbuvir, a fixed-dose combination tablet for the treatment of hepatitis C. Clin. Pharmacokinet., 55, 1337–1351 (2016).
- 14) Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv. Drug Deliv. Rev., 55, 3–29 (2003).
- 15) Company BMS. Sunvepra Capsules (asunaprevir) Japanese Prescribing Information. (2014).
- 16) Hebert MF, Roberts JP, Prueksaritanont T, Benet LZ. Bioavailability of cyclosporine with concomitant rifampin administration is markedly less than predicted by hepatic enzyme induction. Clin. Pharmacol. Ther., 52, 453–457 (1992).
- 17) Reddy MB, Chen Y, Haznedar JO, Fretland J, Blotner S, Smith P, Tran JQ. Impact of low-dose ritonavir on danoprevir pharmacokinetics: results of computer-based simulations and a clinical drug-drug interaction study. Clin. Pharmacokinet., 51, 457–465 (2012).
- 18) Brennan BJ, Poirier A, Moreira S, Morcos PN, Goelzer P, Portmann R, Asthappan J, Funk C, Smith PF. Characterization of the transmembrane transport and absolute bioavailability of the HCV protease inhibitor danoprevir. Clin. Pharmacokinet., 54, 537–549 (2015).
- 19) German P, Mathias A, Brainard DM, Kearney BP. Drug-drug interaction profile of the fixed-dose combination tablet regimen Ledipasvir/Sofosbuvir. Clin. Pharmacokinet., 57, 1369–1383 (2018).
- 20) Ouwerkerk-Mahadevan S, Snoeys J, Peeters M, Beumont-Mauviel M, Simion A. Drug-drug interactions with the NS3/4A protease inhibitor Simeprevir. Clin. Pharmacokinet., 55, 197–208 (2016).
- 21) Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. U.S.A., 84, 7735–7738 (1987).
- 22) Cordon-Cardo C, O’Brien JP, Boccia J, Casals D, Bertino JR, Melamed MR. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J. Histochem. Cytochem., 38, 1277–1287 (1990).
- 23) Lin JH, Yamazaki M. Clinical relevance of P-glycoprotein in drug therapy. Drug Metab. Rev., 35, 417–454 (2003).
- 24) Cao X, Yu LX, Barbaciru C, Landowski CP, Shin HC, Gibbs S, Miller HA, Amidon GL, Sun D. Permeability dominates in vivo intestinal absorption of P-gp substrate with high solubility and high permeability. Mol. Pharm., 2, 329–340 (2005).
- 25) Dahan A, Amidon GL. Segmental dependent transport of low permeability compounds along the small intestine due to P-glycoprotein: the role of efflux transport in the oral absorption of BCS class III drugs. Mol. Pharm., 6, 19–28 (2009).
- 26) Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res., 12, 413–420 (1995).
- 27) Weiss J, Becker JP, Haefeli WE. Telaprevir is a substrate and moderate inhibitor of P-glycoprotein, a strong inductor of ABCG2, but not an activator of PXR in vitro. Int. J. Antimicrob. Agents, 43, 184–188 (2014).
- 28) Bleasby K, Castle JC, Roberts CJ, Cheng C, Bailey WJ, Sina JF, Kulkarni AV, Hafey MJ, Evers R, Johnson JM, Ulrich RG, Slatter JG. Expression profiles of 50 xenobiotic transporter genes in humans and pre-clinical species: a resource for investigations into drug disposition. Xenobiotica, 36, 963–988 (2006).
- 29) Katoh M, Suzuyama N, Takeuchi T, Yoshitomi S, Asahi S, Yokoi T. Kinetic analyses for species differences in P-glycoprotein-mediated drug transport. J. Pharm. Sci., 95, 2673–2683 (2006).
- 30) Takeuchi T, Yoshitomi S, Higuchi T, Ikemoto K, Niwa S, Ebihara T, Katoh M, Yokoi T, Asahi S. Establishment and characterization of the transformants stably-expressing MDR1 derived from various animal species in LLC-PK1. Pharm. Res., 23, 1460–1472 (2006).
- 31) Yamasaki Y, Kobayashi K, Okuya F, Kajitani N, Kazuki K, Abe S, Takehara S, Ito S, Ogata S, Uemura T, Ohtsuki S, Minegishi G, Akita H, Chiba K, Oshimura M, Kazuki Y. Characterization of P-glycoprotein humanized mice generated by chromosome engineering technology: its utility for prediction of drug distribution to the brain in humans. Drug Metab. Dispos., 46, 1756–1766 (2018).
- 32) Lin JH, Yamazaki M. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin. Pharmacokinet., 42, 59–98 (2003).
- 33) Eley T, Garimella T, Li W, Bertz RJ. Asunaprevir: a review of preclinical and clinical pharmacokinetics and drug–drug interactions. Clin. Pharmacokinet., 54, 1205–1222 (2015).
- 34) Garg V, van Heeswijk R, Lee JE, Alves K, Nadkarni P, Luo X. Effect of telaprevir on the pharmacokinetics of cyclosporine and tacrolimus. Hepatology, 54, 20–27 (2011).