Review
Chemistry of Fluorinated Carbon Acids: Synthesis, Physicochemical Properties, and Catalysis
2015 Volume 63 Issue 9 Pages 649-662
Details
2015 Volume 63 Issue 9 Pages 649-662
The bis[(trifluoromethyl)sulfonyl]methyl (Tf2CH; Tf=SO2CF3) group is known to be one of the strongest carbon acid functionalities. The acidity of such carbon acids in the gas phase is stronger than that of sulfuric acid. Our recent investigations have demonstrated that this type of carbon acids work as novel acid catalysts. In this paper, recent achievements in carbon acid chemistry by our research group, including synthesis, physicochemical properties, and catalysis, are summarized.
Brønsted acid is one of the most classical catalysts in organic synthesis. In this context, sulfuric acid has occupied a central position for a long time. On the other hand, organic chemists have dedicated substantial efforts to discovering organic acids stronger than sulfuric acid. As a revolutionary study, Haszeldine and Kidd reported trifluoromethanesulfonic acid TfOH (Tf=CF3SO2) as a superacidic organic compound during the mid-1950s.1) The Hammett acidity function H0 of TfOH was determined to be approximately −14.2) This value means that it is more acidic than sulfuric acid (H0=−12).3) Recent calculations have also proposed that the pKa value of TfOH in H2O is −14.2.4) In 1984, Foropoulos and DesMarteau reported research on bis(triflyl)imide Tf2NH, which was another important example of a superacidic compound.5)
Currently, these superacidic organic acids and their conjugate bases are widely used in several fields of chemistry including catalysis, ionic liquids, and material sciences. Structurally similar carbon acids such as Tf2CH2 16,7) and Tf3CH8) also have long histories. However, these carbon (C–H) acids have not been thoroughly investigated. One of the major reasons for this is likely to be the lack of effective synthetic procedures. As an ongoing research program, we are studying methods to synthesize carbon acid derivatives and their applications as synthetically useful reagents and/or organocatalysts. The purpose of this review is to provide an outline of the carbon acid chemistry studies undertaken by our research group.
Although active methylenes such as 1,3-dicarbonyl compounds react with organic/inorganic bases to give synthetically useful carbanion species, their acidities are not sufficient to catalyze typical acid-catalyzed reactions. However, some acidity scales unexpectedly indicated considerable acidity of Tf2CH2 1 (Table 1). For example, the gas-phase acidity GA (or ΔGacid) of sulfuric acid and Tf2CH2 1 was reported to be 302.2 kcal mol−1 and 300.6 kcal mol−1, respectively.9,10) These values support the superacidity of 1 in the gas phase. In addition, the pKa value of 1 in dimethyl sulfoxide (DMSO) (2.1) suggested its markedly strong acidity in the solution phase; the acidity of 1 was slightly weaker than that of sulfuric acid (pKa=1.4), although it worked as a stronger acid compared with CF3CO2H (pKa=3.45).11,12)
| Acid | pKa (DMSO) | GA (kcal mol−1) |
|---|---|---|
| H2SO4 | 1.4 | 302.2 |
| TfOH | 0.3 | 299.5 |
| Tf2NH | — | 286.5 |
| Tf3CH | — | 289.0 |
| Tf2CH2 (1) | 2.1 | 300.6 |
| CF3CO2H | 3.45 | — |
| (PhSO2)2CH2 | 12.25 | — |
| (EtO2C)2CH2 | 16.2 | — |
| TfCH3 | 18.8 | 339.8 |
In terms of such aspects of carbon acid 1 in both the gas and solution phases, we were interested in catalysis of carbon acids bearing the Tf2CH group as an acidic functionality. In the carbon acid structure, it is possible to incorporate a fourth substituent R, which provides some additional functionalities to the acid, on the central carbon atom (Chart 1). In principle, such a molecular design to develop highly effective acid catalysts would be difficult in the cases of the corresponding oxygen and nitrogen acids.
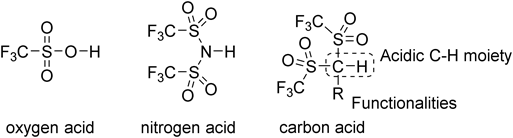
However, it is not easy to synthesize carbon acids with structural diversity.13) For example, Koshar reported the α-alkylation reaction of Tf2CH2 114) (Chart 2). Unfortunately, the desired alkylation products were obtained in merely poor-to-moderate yields since the carbanion derived from 1 was significantly stabilized by two triflyl groups, and its nucleophilicity was not suitable for the subsequent substitution with conventional alkyl halides. As an alternative approach to construct the carbon acid structure, Ishihara and Yamamoto developed an α-triflylation reaction of monosulfones with Tf2O15) (Chart 3). This was fairly effective for the aryl-substituted carbon acids ArCHTf2 including pentafluorophenyl carbon acid 2a,16,17) fluorous-tagged carbon acids,18,19) and chiral derivative 2b.20) In addition, they also found that these types of carbon acids served as Brønsted acid catalysts for several C–C bond-forming reactions. However, in this synthesis, strongly basic and ignitable t-BuLi was necessary for the α-triflylation step. This would narrow the functional group tolerance. Furthermore, troublesome repeating reaction operations were required to obtain the desired carbon acids in pure form.

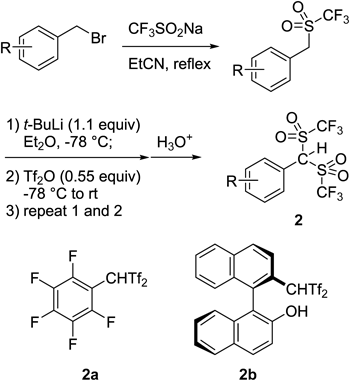
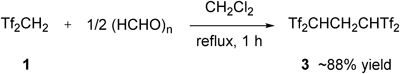
On the basis of such pioneering works, we adopted 1,1,3,3-tetrakis(triflyl)propane (Tf2CHCH2CHTf2) 3 as the first generation of our carbon acid catalysts (Chart 4). This compound was originally reported in 1976 by Koshar and Barber.21) Although Tf2CHCH2CHTf2 3 was easily synthesized in multigram scale by the 2 : 1 reaction of Tf2CH2 122) with paraformaldehyde, followed by recrystallization from chlorobenzene, the applications and detailed physicochemical properties had not been described.23)
According to Koshar’s procedure, we obtained Tf2CHCH2CHTf2 3 as colorless crystals. Interestingly, this acid did not show deliquescent and fuming properties under an air atmosphere. The X-ray crystallographic structure of 3 is shown in Chart 5.24) The acidic C–H moieties are located inside the molecular structure, while fluorine and oxygen atoms occupy the outside. The gas-phase acidity of 3 was shown to be 290.2 kcal mol−1 using the Fourier transform-ion cyclotron resonance (FT-ICR) technique.25)
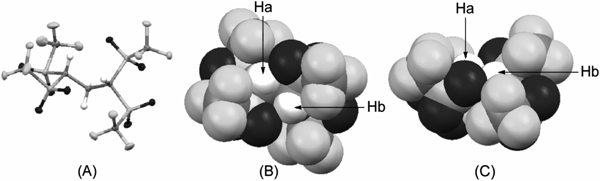
(A) ORTEP drawing; (B) space-filling model, top view; (C) space-filling model, side view.
With Tf2CHCH2CHTf2 3 in our hands, its catalysis was evaluated in the vinylogous Mukaiyama–Michael (VMM) reaction of mesityl oxide 4a with 2-silyloxyfuran26) (Table 2). In the presence of a catalytic amount of TfOH and Tf2NH, the reaction gave the desired product 5a in only low yields. In contrast, when using 0.25 mol% of 3 instead of TfOH and Tf2NH, 5a was obtained in 88% yield after acid treatment. The loading of 3 could be reduced to 0.05 mol% without significant loss of the product yield. This VMM reaction did not occur in the absence of acid catalysts. Moreover, Lewis acids such as Me3Al required substoichiometric loading to achieve acceptable consumption of the substrates.
 | |||
|---|---|---|---|
| Entry | Acid (mol%) | Temp. (°C) | Yielda) (%) |
| 1 | TfOH (0.25) | −78 | 7 |
| 2 | Tf2NH (0.25) | −78 | 7 |
| 3 | Tf2CHCH2CHTf2 3 (0.25) | −78 | 88 |
| 4 | Tf2CHCH2CHTf2 3 (0.05) | −24 | 87 |
| 5 | Tf2CH2 1 (1.0) | −78 | 0 |
| 6 | Tf2CHCH3 (1.0) | −78 | 7 |
| 7 | Tf2CHC6F5 2a (0.05) | rt | 36 |
| 8 | Me3Al (40) | −78 | 64 |
| 9 | None | rt | NRb) |
a) Isolated yield. b) No reaction.
Under the optimized conditions using Tf2CHCH2CHTf2 3, several α,β-enones smoothly produced the desired VMM products in good-to-excellent yields (Chart 6). Upon treatment with 3-bromo-2-silyloxyfuran, β-alkylated enones 4 gave the corresponding VMM products 5 in a highly anti-selective manner. Importantly, this carbon acid-induced reaction achieved C–C bond formation between sterically hindered substrates.

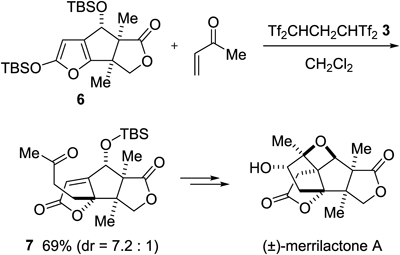
As an elegant extension of our VMM chemistry, Zhai and colleagues used it as a key step for the synthesis of (±)-merrilactone A27) (Chart 7). In the presence of a catalytic amount of 3, the reaction of tricyclic silyloxyfuran 6 with methyl vinyl ketone gave the VMM product 7 in 69% yield. They also found that the uses of typical Lewis acids instead of 3 were less effective for this transformation.
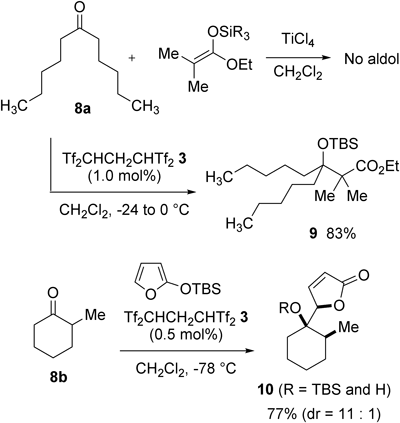
The notable catalysis of Tf2CHCH2CHTf2 3 was observed not only in the VMM reaction but also in the Mukaiyama aldol reaction.28) Rhodes pointed out that the reaction of sterically bulky undecan-6-one 8a with a ketene silyl acetal did not yield the corresponding aldol product under classical titanium tetrachloride (TiCl4)-mediated conditions29) (Chart 8). On the other hand, only 1.0 mol% of 3 was sufficient to complete this reaction, giving rise to 9. Likewise, tetrasulfone 3 was successfully applied to the diastereoselective vinylogous Mukaiyama aldol (VMA) reaction of α-alkylated cyclohexanones such as 8b using 2-silyloxyfurans.30)
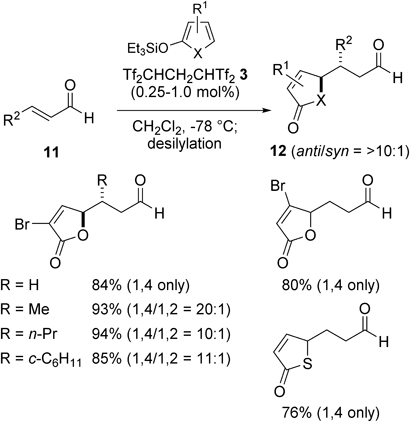
Surprisingly, a switching of the regioselectivity between the carbon acid-catalyzed conditions and the typical Lewis acid-mediated conditions was observed in the reaction of α,β-unsaturated aldehydes with 2-silyloxyfurans.31) De Lera and colleagues demonstrated that the reaction of aldehydes 11 with 3-bromo-2-silyloxyfuran in the presence of BF3·OEt2 selectively gave the corresponding 1,2-adducts in good yields.32) In contrast, the unexpected 1,4-adducts 12 were selectively obtained by using Tf2CHCH2CHTf2 3 instead of BF3·OEt2 (Chart 9). A high level of 1,4-selectivity was also observed in the reactions using 2-silyloxythiophene.
As a plausible reaction pathway for the Tf2CHCH2CHTf2-induced Mukaiyama aldol and related reactions, we proposed the in-situ generation of silyl alkanide intermediate A as a catalytically active species (Chart 10). That is, initial protonation of ketene silyl acetals by carbon acid 3 and the subsequent silyl transfer reaction produced silyl alkanide A, which shows a high level of silylating activity due to the intramolecular strain (I-strain) between the trialkylsilyl group and sterically bulky gem-triflylated alkanide moiety.33,34) This species activates the carbonyl substrates via the O-silylation reaction, and then nucleophilic attack of unreacted ketene silyl acetals on the resulting silyl carboxonium B forms the C–C bond (formation of C). Furthermore, the silyl transfer reaction from intermediate C to unreacted carbonyl substrates regenerates intermediate B along with the formation of aldol (or Michael) product D.35) If highly Lewis acidic species A is inactivated by a trace amount of water existing in the reaction system, a self-repairing pathway through the protonation of ketene silyl acetals by Tf2CHCH2CHTf2 3 will bring about the regeneration of catalytically active A.36) Although our efforts to observe intermediate A failed, some experimental results supported this plausible mechanism. First, in 1H- and 13C-NMR studies using a 1 : 1 mixture of mesityl oxide 4a and 3, considerable shifts of each NMR signal were not observed. Second, the VMM reaction was not impeded by the addition of 2,6-di-tert-butylpyridine as a specific proton scavenger. These data imply that electrophilic activation of the carbonyl substrates through protonation or hydrogen bonding by carbon acid 3 is not a major activation mode for the present C–C bond-forming reactions.

Compared with TfOH and Tf2NH, Tf2CHCH2CHTf2 3 could be used as more powerful acid catalyst for the Mukaiyama aldol and related reactions. This finding encouraged us to develop practical synthetic strategies for novel carbon acid derivatives. To synthesize Tf2CH-based carbon acids with structural diversity, both synthetic reactions and purification methods should be considered. In most of the literature describing carbon acid synthesis, carbon acids were isolated by distillation, sublimation, or recrystallization. This indicates the difficulty in chromatographic purification of carbon acids.37) In other words, practical synthetic strategies for this type of carbon acids must meet specific requirements. First, essentially quantitative formation of carbon acids is required for the reaction itself. Second, side products formed by carbon acid synthesis must be easily removable using simple purification techniques. Third, carbon acid-incorporating reactions should be applicable to structurally complex substrates.
To develop a satisfactory methodology, we were interested in 1,1-bis(triflyl)ethylene Tf2C=CH2 13,38) which was proposed as a reactive intermediate in Koshar’s synthesis of Tf2CHCH2CHTf2 321) (Chart 11). If Tf2C=CH2 13 generated in situ reacts with neutral nucleophiles, the corresponding carbon acids will be formed as adducts. After many attempts, we developed three different reactions for the generation of 13: 1) retro-Michael reaction of Tf2CHCH2CHTf2 3; 2) self-promoting condensation reaction of Tf2CH2 1 and formaldehyde; and 3) decomposition reaction of zwitterion 14a.

Although our first-generation catalyst Tf2CHCH2CHTf2 3 was a bench-stable chemical as crystals, it provided an equilibrium mixture of Tf2CH2 1/Tf2C=CH2 13 and Tf2CHCH2CHTf2 3 in diluted solutions in several organic solvents at higher temperature. As shown in Chart 12, 1H-NMR analysis of a 0.01 M solution of Tf2CHCH2CHTf2 3 in CDCl3 at 40°C revealed this equilibrium profile (Keq=3.21×10−3). A similar NMR study of a 0.01 M solution in CD3CN revealed rapid, complete dissociation of 3, giving rise to Tf2CH2 1 and Tf2C=CH2 13.

Squares, Tf2CHCH2CHTf2 3; diamonds, Tf2CH2 1; triangles, Tf2C=CH2 13.
In actuality, Tf2CHCH2CHTf2 3 worked as an effective reagent to incorporate the 2,2-bis(triflyl)ethyl group(s) into electronically rich arene frameworks.39) When 2,6-xylenol was treated with 3 in acetonitrile at room temperature, the desired carbon acid 15a was obtained in 94% yield after bulb-to-bulb distillation using a Kugelrohr oven (Chart 13). Meanwhile, Tf2CH2 1 was recovered in 90% yield. The three-component reaction of 2,6-xylenol, 1, and paraformaldehyde yielded carbon acid 15a as well as inseparable diarylmethane as a side product. In the latter case, relatively slow formation of Tf2C=CH2 13 would result in competitive formation of the side product. Some carbon acids with high boiling points were also synthesized and isolated on the basis of this reaction. For example, acceptably pure 2,6-diphenyl derivative 15b was obtained in 97% yield by the reaction of 2,6-diphenylphenol with 3, followed by the removal of Tf2CH2 1 using a Kugelrohr oven.

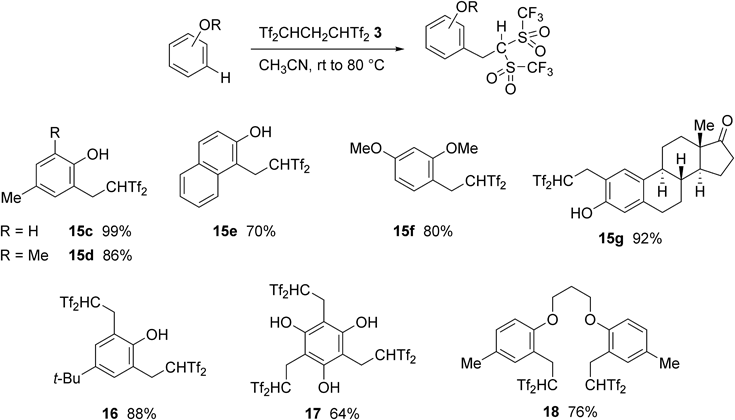
Selected results of the 2,2-bis(triflyl)ethylating reaction of electronically rich arenes such as phenols and aryl ethers with Tf2CHCH2CHTf2 3 are summarized in Chart 14. Interestingly, the reaction of 4-substituted phenol with 1 equiv. of 3 gave the monosubstituted product 15c in 99% yield. Under similar conditions, the 1 : 2 reaction of 4-tert-butylphenol with 3 predominantly gave disubstituted product 16. Triple-carbon acid 17 was also obtained by the reaction of benzene-1,3,5-triol with 3 equiv. of 3. The present methodology was markedly effective for the carbon acid introduction of structurally complex aromatics; e.g., estrone-based carbon acid 15g and bis-carbon acid 18 were synthesized in good yields, respectively.
We found that 1,3-dicarbonyl compounds and succinic imide served as acceptable reaction partners.40) As shown in Chart 15, highly functionalized carbon acids bearing the Tf2CH group were easily obtained using our method.
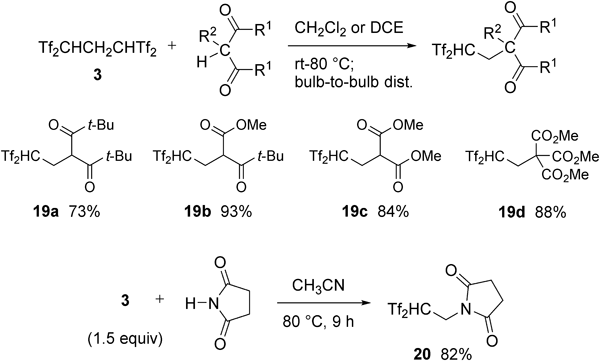
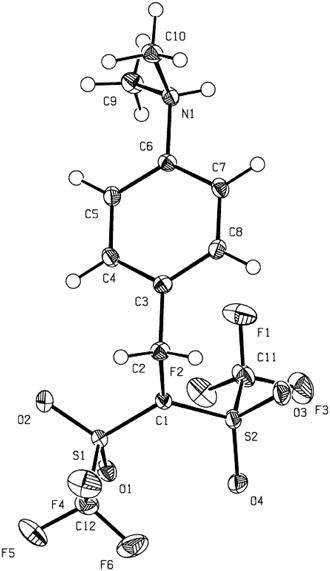
By treating aniline derivatives with Tf2CHCH2CHTf2 3, unusual zwitterions bearing both stabilized carbanion and ammonium moieties in the molecular structure were isolated41) (Chart 16). Their zwitterionic structures were fully confirmed by X-ray crystallography and/or NMR analysis. When N,N-dimethylaniline was treated with an equimolar amount of 3 in acetonitrile for 3 h at 80°C, the para-substituted product 21a was quantitatively formed as white precipitates after evaporation to remove the solvent and Tf2CH2 1. The zwitterion 21a was isolated in 95% yield by washing the precipitates with chloroform. As shown in Chart 17, X-ray crystallographic analysis of 21a fully supported the sp2-hybridizing nature of anionic C1 because the bond angles around the C1 were 120.5° (C2–C1–S1), 120.3° (S1–C1–S2), and 119.2° (S2–C1–C2), respectively. At the same time, the bond angles around the cationic N1 (114.3° for C6–N1–C9, 110.3° for C9–N1–C10, and 110.4° for C10–N1–C6) suggested its tetrahedral structure. Furthermore, the hydrogen on the N1 atom was found from the crystallographic data. Likewise, several N-substituted anilines were smoothly converted to the corresponding zwitterions 21b–i in good-to-excellent yields.

Tf2CHCH2CHTf2 3 itself could be prepared by the 2 : 1 reaction of Tf2CH2 1 and paraformaldehyde in multigram scale. This suggested the possibility of the self-promoting condensation reaction of Tf2CH2 1 and paraformaldehyde as another choice for the generation of Tf2C=CH2 13. However, compared with our methodology using Tf2CHCH2CHTf2 3, the reaction rate of this condensation was significantly slow, and a 1 : 1 reaction of 1 with paraformaldehyde gave only an equilibrium mixture of Tf2CH2 1, Tf2C=CH2 13, and Tf2CHCH2CHTf2 3. Well-considered choices of nucleophiles were obviously required in this case.
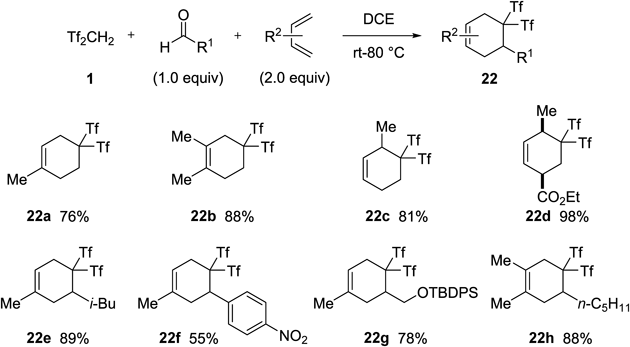
First, we found the Diels–Alder reaction of Tf2C=CH2 13 with 1,3-dienes42) (Chart 18). By mixing 1, paraformaldehyde, and isoprene, the desired three-component reaction including the in-situ generation of 13 followed by cycloaddition proceeded to give gem-bis(triflyl)cyclohexene 22a in 76% yield. A similar transformation occurred from Tf2CHCH2CHTf2 3 with isoprene, although the yield of 22a (61%) was slightly decreased due to the competitive polymerization of isoprene. It should be noted that only the para-adduct was obtained in a regiospecific manner. Several 1,3-dienes were also subjected to this three-component synthesis. In particular, the reaction using less reactive ethyl sorbate as the diene component gave the desired cyclohexene 22d in excellent yield. Under similar conditions, not only paraformaldehyde but also some aromatic and aliphatic aldehydes could be used as effective reaction substrates.
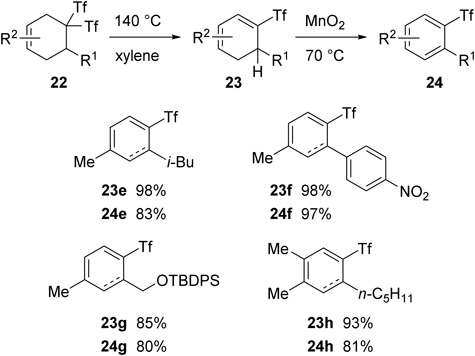
The gem-bis(trifly)cyclohexenes thus obtained were easily converted into the corresponding triflylarenes (Chart 19). For example, 2,4-dialkylated triflylbenzenes 24e–h were obtained in good overall yields from the cyclohexenes through the thermal elimination of trifluoromethanesulfinic acid and subsequent aromatization using Corey’s procedure.43,44)
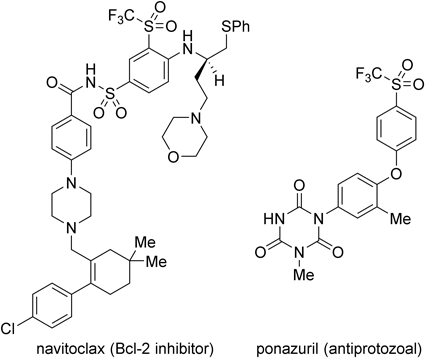
Recently, triflylated aromatics including navitoclax (ABT-263) and ponazuril have attracted a great deal of attention in medicinal chemistry (Chart 20). Such highly substituted triflylarenes were traditionally synthesized through a combination of aromatic electrophilic substitution reactions. In our present synthesis, the relative configuration of each substituent including the triflyl group on the benzene ring was perfectly controlled in the Diels–Alder reaction step. The reaction provided a new entryway to synthesize highly substituted triflylarenes without the formation of undesired regioisomers.
That success inspired us to explore stable, isolable 1,1-bis(triflyl)alkenes bearing substituents on the sulfone β-carbon. In fact, Koshar pointed out the difficulties in isolating Tf2C=CH2 13 and its handling.37) Zhu also reported that β-phenylated 1,1-bis(triflyl)alkene was easily hydrolyzed by moisture under an air atmosphere to give Tf2CH2 1 and benzaldehyde.45) If stable, isolable 1,1-bis(triflyl)alkenes are found, systematic investigations of their reactions with a wide range of nucleophiles including strongly basic and/or anionic nucleophiles will be possible.
To improve the stability of the 1,1-bis(triflyl)alkenes, we focused on 1,1-bis(triflyl)alkadienes derived from Tf2CH2 1 and α,β-unsaturated aldehydes. Here the alkadienes would show higher stability to water because their thermodynamic stability possibly improved with a sterically less hindered π-conjugation system. At the same time, an improvement in their kinetic stability could also be considerable (Chart 21).

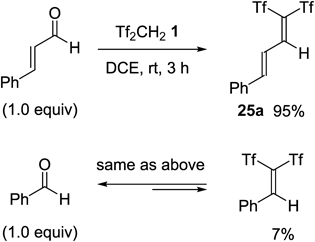
As a result, we successfully isolated the 1,1-bis(triflyl)alkadienes by reaction with cinnamaldehyde derivatives.46,47) The reaction of cinnamaldehyde with Tf2CH2 1 in 1,2-dichloroethane (DCE) gave the desired alkadiene 25a in 95% yield. This product was obtained as yellow crystals, and we did not observe their decomposition under an air atmosphere during a period of at least several months. In contrast, a similar reaction of benzaldehyde gave the alkene product in only 7% yield. This sharp contrast in the reaction outcomes indicated the significantly improved stability of 25a (Chart 22).
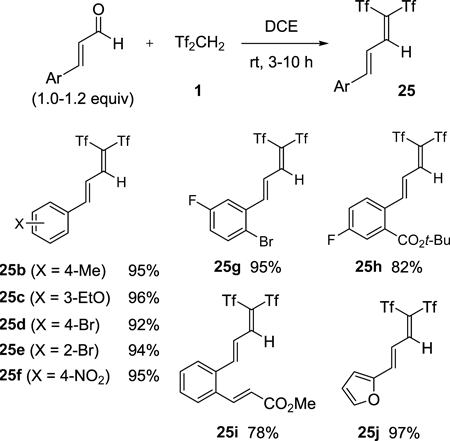
Moreover, substituted cinnamaldehydes smoothly gave 1,1-bis(triflyl)alkadienes 25 in good-to-excellent yields (Chart 23). This condensation reaction proceeded without touching acid-sensitive functionalities such as the tert-butyl ester, unsaturated ester, and furan moieties.
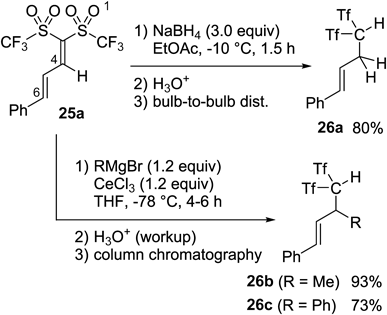
The obtained alkadienes 25 could be used as effective building blocks for carbon acids through reactions with several anionic nucleophiles (Chart 24). To give examples, the selective 1,4-reduction of 25a was achieved by treatment with NaBH4 in EtOAc at −78°C, and cinnamyl carbon acids 26a were isolated in 80% yield after bulb-to-bulb distillation.44) Likewise, the reaction of 25a with organocerium reagents prepared from Grignard reagents and anhydrous CeCl3 produced the β-branched carbon acids 26b and c.47) In this nucleophilic alkylation chemistry, we had to employ column chromatography on neutral silica gel to separate the desired β-alkylated carbon acids and a small amount of impurity mainly containing γ-alkylation products. After defined experiments, it was found that carbon acids were eluted as the corresponding Ca2+ salts during usual column chromatography. The free carbon acids were obtained by reacidification of the Ca2+ salts with 10% hydrochloric acid.
The in-situ generation of Tf2C=CH2 13 using Tf2CHCH2CHTf2 3 or Tf2CH2 1/paraformaldehyde became the carbon acid synthesis. Although the reaction of neutral nucleophiles such as electron-rich aromatics and active methylenes with Tf2CHCH2CHTf2 3 resulted in the clean formation of carbon acids, Tf2CH2 1 was also recovered as a side product. In order to reuse this Tf2CH2 1 for the same purpose, this compound should be converted into the tetrasulfone 3 since the self-promoting condensation reaction of 1 and paraformaldehyde was relatively slow and acceptable nucleophiles in this case were significantly limited. Such problems to be resolved led us to make further efforts for developing a more effective reagent. We took note of pyridinium type zwitterions as the third entry of Tf2C=CH2 precursors. In 1976, Koshar and co-workers reported that the reaction of Tf2CH2 1 and paraformaldehyde gave a mixture of 13 and Tf2CHCH3 in a ratio of 42 : 58, and the subsequent treatment of this mixture with a large excess of pyridine gave 14b in 41% yield38) (Chart 25). In that report, they proposed its zwitterionic structure bearing an unusual carbanion moiety on the basis of elemental analysis alone. We reexamined this reaction. After several attempts, we found that by mixing Tf2CH2 1, paraformaldehyde, and pyridine for 4 h at 60°C, 14b was formed in quantitative yield. The molecular structure of 14b was fully confirmed in X-ray crystallographic analysis.
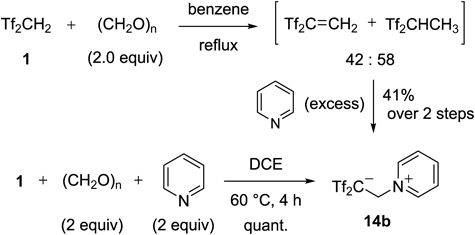
Our three-component reaction could be applied to several nitrogen-containing heterocycles (Chart 26). For example, the reactions of 2-substituted pyridines gave the corresponding zwitterions 14a–g. Under similar conditions, quinolinium 14h, oxazolium 14i, and imidazolium 14j were isolated in essentially quantitative yields, respectively.
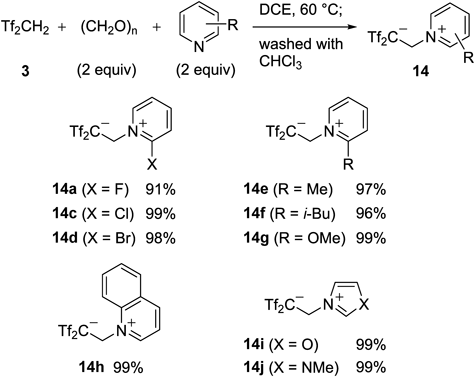
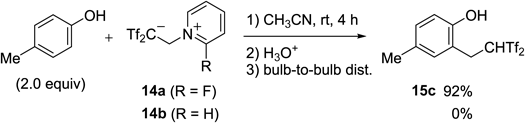
Among these, 2-fluoropyridinum 14a could be used as a markedly effective reagent for the in-situ generation of Tf2C=CH2 13. The reaction of 14a with p-cresol rapidly gave the desired carbon acid 15c in 92% yield, whereas nonfluorinated pyridinium 14b did not give 15c under the same conditions (Chart 27). This reagent has the following advantages: 1) a stable crystalline compound without deliquescent properties; 2) significantly fast rate for the formation of Tf2C=CH2 13 in several organic solvents; and 3) low boiling point of 2-fluoropyridine, which was formed as a side product during the 2,2-bis(triflyl)ethylation reaction, and its remarkably weak basicity (pKaH in H2O=−0.44).48)
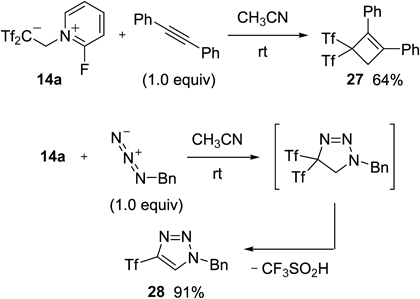
Recently, Alcaide and Almendros have reported interesting cycloaddition chemistry using our 2-fluoropyridinium 14a (Chart 28). For example, Tf2C=CH2 13 generated in situ from 14a smoothly reacted with the internal alkyne to give gem-bis(triflyl)cyclobutene 27.49) Likewise, the reaction with benzyl azide gave triflyl-1,2,3-triazole 28 through (3+2) cycloaddition, followed by desulfinylation under mild conditions.50) These results clearly demonstrated an advantage of our zwitterion in the synthesis of not only carbon acids but also triflylated chemicals.
We also evaluated the acidity of several carbon acid derivatives in both gas and solution phases. Selected data are summarized in Chart 29. In a series of phenol-derived carbon acids 15d, 16, and 17, the pKa values in DMSO solution were determined to be 2.3, 2.1, and 2.0, respectively,39) using an electrochemical method.51,52) Compared with the pKa values of Tf2CH2 1 (2.1) and phenol (18) in DMSO solutions,11) the values measured in our acids suggested that the major acidic functionality was the Tf2CH group rather than the phenolic hydroxyl group. On the other hand, the pKa value of acidic zwitterion 21a was determined to be 2.4. This zwitterion was a slightly weaker acid than carbon acids. Interestingly, by increasing in the number of the Tf2CH groups on the benzene ring, the acidity was slightly enhanced. A key structural unit to intensify the acidity was also found in comparisons of the GA. Experimentally determined GA values of 15d and 16 were 294.8 kcal mol−1 and 291.5 kcal mol−1, respectively.25) Unfortunately, the GA value of triple-carbon acid 17 was not determined in this experimental manner, but it was predicted to be 278.9 kcal mol−1 by a high-level density functional theory (DFT) calculation.53) In this computation, it was revealed that the conjugate base of 17 was highly stabilized by two hydrogen bonds between phenolic hydroxyl groups and sulfonic oxygens. The proximate hydroxyl groups on the benzene ring played an important role in achieving notable enhancement of the acidity.
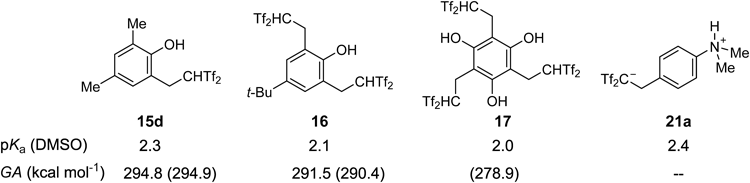
Calculated GA values (B3LYP/6-311++G** level) are shown in parentheses.
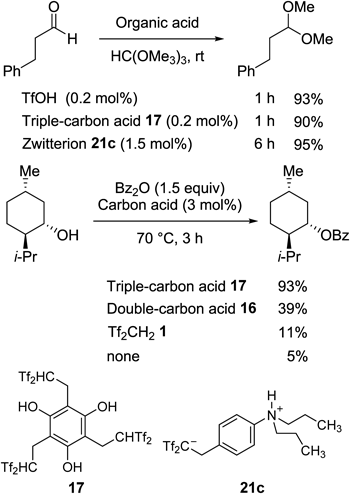
Such phenol-derived carbon acids and acidic zwitterions could be used as real Brønsted acid catalysts. For example, both triple-carbon acid 17 and N,N-dipropylated zwitterion 21c nicely catalyzed the acetal-forming reaction of 3-phenylpropanal, which was a typical example of Brønsted acid-catalyzed reactions39) (Chart 30). Moreover, triple-carbon acid 17 served as an effective catalyst for the esterification reaction of menthol. It is known that this reaction is not catalyzed by the strongly acidic polymer Nafion SAC-13.17)
Next, we examined the utility of the carbon acid derivatives in the Mukaiyama aldol-type reactions. As mentioned above, our carbon acids could be used as real Brønsted acid catalysts. However, the activation of carbonyl substrates through protonation or hydrogen bonding by carbon acids would not stand out compared with other common acids (Chart 31). On the other hand, as shown in the Tf2CHCH2CHTf2-induced reactions, silicon Lewis acids derived from carbon acids would become a rational approach for notably strong activation of the carbonyl substrates. Such reaction systems were the formal Brønsted acid catalysis by carbon acids. In particular, the relationship between the molecular structures of a series of carbon acid derivatives and their catalytic performances was an interesting aspect.

To provide insight into the structure–catalyst activity relationship, we evaluated the catalyst performance of the phenol-derived carbon acids in the VMA reaction of cyclohexanone 8c with 2-silyloxyfuran39) (Table 3). In this reaction, our first-generation catalyst Tf2CHCH2CHTf2 3 had shown higher efficiency than TfOH and Tf2NH (entry 1 vs. entries 2, 3). The use of monocarbon acid 15a with a carbon acid moiety at the para position to the hydroxyl group instead of tetrasulfone 3 did not show good consumption of 8c and acceptable isolated yield of 29, whereas carbon acid 15c gave a better yield of 29 under similar conditions (entries 4, 5). Notably, double-carbon acid 16 and triple-carbon acid 17 were highly effective for this transformation (entries 6, 7). Under optimized conditions, only 0.05 mol% loading of 17 was sufficient to complete the VMA reaction (entry 8).
 | ||||
|---|---|---|---|---|
| Entry | Organic acid (mol%) | Temp. (°C) | Time (h) | Yield of 29a) (%) |
| 1 | Tf2CHCH2CHTf2 3 (0.5) | −24 | 2.5 | 82 |
| 2 | TfOH (0.5) | −24 | 2.5 | 25 |
| 3 | Tf2NH (0.5) | −24 | 2.5 | 63 |
| 4 | 15a (1.0) | rt | 2 | 30 |
| 5 | 15c (1.0) | rt | 2 | 51 |
| 6 | 16 (1.0) | rt | 2 | 82 |
| 7 | 17 (1.0) | rt | 2 | 83 |
| 8 | 17 (0.05) | rt | 2 | 83 |
a) Combined yield of 29-H (R=H) and 29-Si (R=TBS).
These results confirmed that triple-carbon acid 17 showed a high level of performance as both real and formal Brønsted acid catalysts. In addition, the higher thermal stability of 17 over Tf2CHCH2CHTf2 3 in several solvents was an important point in its catalyst usage. As an example to illustrate the distinguishing features of 17, we found the unusual olefination reaction of lactones using ketene silyl acetals.54) In general, the reactions of carboxylic acid derivatives with silicon enolates give the corresponding Claisen condensation product E55,56) (Chart 32). In similar reactions, silyl acetal D, which is a simple adduct of the silicon enolate to the starting ester, was often isolated.57,58) Furthermore, a few examples demonstrated the olefination reaction giving rise to vinyl ether F.59)

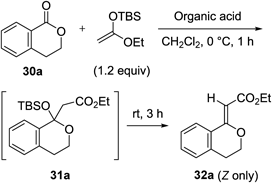
When isochroman-1-one 30a was treated with a ketene silyl acetal in the presence of 1 mol% of triple-carbon acid 17 at 0°C, formation of the adduct 31a was observed in TLC analysis (Table 4, entry 1). Interestingly, silyl acetal 31a was rapidly converted to vinyl ether 32a by additional stirring at room temperature. In this case, vinyl ether 32a was isolated in 51% yield along with the recovery of 31a. The uses of other acids such as TfOH, Tf2NH, and Tf2CHC6F5 2a instead of 17 resulted in poor conversion of 31a (entries 2–4). Furthermore, in the case of Tf2CHCH2CHTf2 3, higher loading was required to obtain essentially the same result (entry 5). After optimization of the reaction conditions with triple-carbon acid 17, we finally obtained (Z)-vinyl ether 32a in 85% yield (entry 7).
 | ||
|---|---|---|
| Entry | Organic acid (mol%) | Yielda) (%) |
| 1 | Carbon acid 17 (1.0) | 51 |
| 2 | TfOH (4.0) | 3b) |
| 3 | Tf2NH (4.0) | 25 |
| 4 | Tf2CHC6F5 2a (4.0) | 30b) |
| 5 | Tf2CHCH2CHTf2 3 (4.0) | 53 |
| 6 | Carbon acid 17 (2.0) | 72 |
| 7c) | Carbon acid 17 (2.0) | 85 |
a) Isolated yield. b) Based 1H-NMR analysis of crude mixture. c) 2.0 eq of ketene silyl acetal.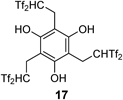
As shown in Chart 33, the present olefination reaction of lactone carbonyls could be applied to several arene-fused lactones 30. Importantly, the decomposition of the resulting vinyl ether moiety in products 32 was not observed during the reaction.

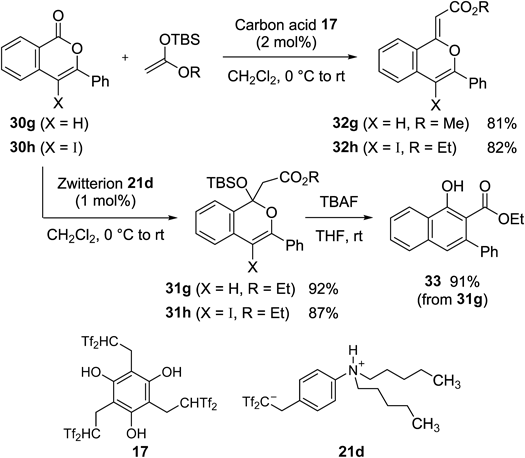
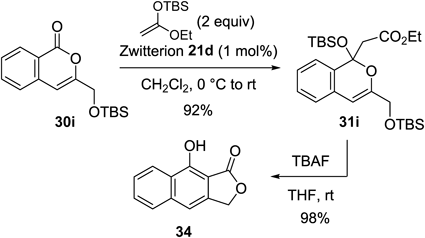
Isocoumarins 30g and h, which had vinyl ester functionalities, were also converted to the corresponding olefination products 32g and h, respectively, without any problems (Chart 34). In contrast, less acidic zwitterion 21d achieved selective formation of silyl acetals 31g and h.60) The later addition chemistry provided a useful synthetic strategy for polysubstituted naphthalenes. For example, by treating the adduct 31g with tetrabutylammonium fluoride (TBAF), 1,2,3-trisubstitued naphthalene 33 was obtained in 91% yield. As another demonstration, we successfully carried out the zwitterion-induced addition reaction, followed by treatment with fluoride to synthesize tricyclic compound 34 (Chart 35).
Chemistry of the strongly acidic carbon acids including Tf2CH-substituted compounds has been extremely limited for many years, even though such acids were discovered much earlier (the mid-1950s). In order to study carbon acids, we first developed an effective synthetic methodology via the in-situ generation of highly electrophilic Tf2C=CH2 13, followed by addition reactions with neutral nucleophiles. On the basis of this, carbon acids with a wide range of structural diversity were easily synthesized. Our studies provided systematic synthesis of a series of carbon acids. The relationship between the molecular structures and their catalyst performances was also defined. In addition, we found that some zwitterions bearing a gem-bis(triflyl)ated carbanion moiety were stable and isolable chemical species.
In several fields of chemistry, strong but easily handled acids have great potential demand. We are continuing systematic research on carbon acid derivatives aimed at the development of highly effective organic acid catalysts.
The author expresses sincere appreciation to Professor Emeritus Takeo Taguchi (Tokyo University of Pharmacy and Life Sciences, TUPLS) and Professor Takashi Matsumoto (TUPLS) for their continued support and useful discussions. The author is also grateful to Professor Emeritus Fumiyo Kusu (TUPLS), Dr. Akira Kotani (TUPLS), Professor Emeritus Masaaki Mishima (Kyushu University), and Professor Emeritus Takaaki Sonoda (Kyushu University) for acidity measurements. The research reported here was made possible by the contributions of several collaborators, including students whose names are acknowledged in the publications cited. Central Glass Co., Ltd. kindly provided us Tf2CH2 1 as the gift. The present work was supported in part by a Grant-in-Aid for Young Scientists (B) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, a Grant-in-Aid for Scientific Research (C) and for Innovative Area “Advanced Molecular Transformation by Organocatalysis” from the Japan Society for the Promotion of Science, as well as Grants from Mitsubishi Gas Chemical Co., Ltd., Asahi Glass Foundation, Kurata Memorial Hitachi Science and Technology Foundation, and Hoansha Foundation.
The author declares no conflict of interest.