Measurements
1H-NMR and EPR Spectra of 1–6The purity of the ibuprofen–nitroxides 1–5 was determined by 1H-NMR and EPR spectroscopies (Figs. S1–S11). In 1H-NMR spectroscopy the assignment was achieved as a hydroxylamine by adding excess of hydrazobenzene. EPR spectra of ibuprofen–nitroxides 3–5 in dimethyl sulfoxide (DMSO) show triplet patterns due to 14N hyperfine splitting, aN=1.43–1.53 mT, and g-values of about 2.006, similar to those for tetramethyl-linked 1 and 2 (Figs. S6–S11). EPR line-widths of nitroxides with tetramethyl groups 1, 2 and 6 were 0.086–0.096 mT and those of nitroxides with the more sterically hindered groups 3, 4 and 5 were broad, 0.103–0.198 mT18) (Table 1). DMSO was used as solvent for the measurement of the reduction kinetics.
Table 1. Values of
g-Factors and Nitrogen
hfc Constants
aN (mT) and Line Width (mT) of Six Nitroxides (2 mM in DMSO)
| Compound | g | aN (mT) | Line width (mT) |
|---|
| 1 | 2.0066 | 1.56 | 0.096 |
| 2 | 2.0069 | 1.47 | 0.086 |
| 3 | 2.0060 | 1.53 | 0.198 |
| 4 | 2.0063 | 1.46 | 0.141 |
| 5 | 2.0068 | 1.43 | 0.103 |
| 6 | 2.0069 | 1.47 | 0.088 |
Partition coefficients (log Po/w) of nitroxides 1–6 between n-octanol and phosphate buffer solution (PBS) (0.1 M, pH 7.4) were measured by EPR spectroscopy and compared.7–9) Since ibuprofen–nitroxides were insoluble in PBS due to their lipophilicity, each nitroxide was added in a mixture of 1 mL of n-octanol and 1 mL of PBS (0.1 M, pH 7.4), and the resultant mixture was mixed vigorously for 1 h, and then the mixture was centrifuged at 3000 rpm for 5 min. Both n-octanol and PBS were subjected to the EPR measurement. Due to the lipophilicity of the ibuprophen–nitroxides, nitroxide-radicals in compounds 2–5 were no detectable in PBS. Log Po/w of 1 was also 3.49, very lipophilic.
Reduction Rate Constants, k (M−1s−1) for 200-Folds Excess AsAThe rates of reduction for nitroxides 1–5 were studied under pseudo-first-order conditions using a 200-fold excess of AsA in DMSO. Second-order rate constants, k were obtained by monitoring the decay of the low-field EPR peak height of nitroxides/peak height of the Mn marker from 2 min to 10 min after reaction at 295 K (Fig. 2, Table 2).
Table 2. Second-Order Rate Constants,
k (M
−1s
−1), for Initial Rates of Reduction of Four Nitroxides (20 µM) with 200-Fold Excess of AsA at 295 K, Based on the
Fig. 2 (Mean±S.D.,
n=3)
| Compound | k (M−1s−1) | R2 |
|---|
| 1 | 0.42±0.07 | 0.9998 |
| 2 | 0.10±0.05 | 0.9815 |
| 3 | 0.17±0.06 | 0.9753 |
| 6 | 0.09±0.02 | 0.9787 |
The reduction reaction of five ibuprofen–nitroxides and 3-hydroxymethyl-2,2,5,5-tetramethylpyrrolidin-1-oxyl (6) with 200-fold excess of AsA, showed that the sterically shielded nitroxides 4 and 5, linked with tetraethyl groups, were scarcely subject to the reduction. The hybrid compounds comprised of ibuprofen and the more reactive six-membered ring compounds (1, 3) or the tetramethyl group-linked five-membered ring compounds (2, 6) were subjected to the reduction, and had the order 1>3>2>6. Rate constant was calculated about 1, 2, 3, and 6 which showed the appropriate decay curve. Tetramethylpiperidine nitroxide 1 was 2.47 times faster than dicyclohexylpiperidine nitroxide 3, which was 1.70 times faster than tetramethylpyroline nitroxide 2, which in turn was 1.11 times faster than tetramethylpyrrolidine nitroxide 6. It was assumed that the six-membered ring nitroxides 1 and 3 are ready to react with AsA compared to the five-membered ring nitroxide because they take the same chair-form. The decay by the reduction of 3 was variable and R2 was low because of the EPR spectrum was unstable and signal width of the splitting pattern was broad.
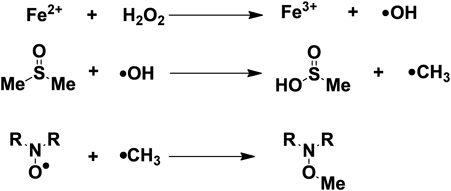
Reduction Rate Constants, k (M−1s−1) for 200-Fold Excess Methyl Radicals Produced by Fenton’s ReagentAs the reaction of AsA with the sterically shielded nitroxides 4 and 5 was limited, a more reactive radical was necessary to obtain more exact rate constants. One such readily preparable radical is the methyl radical, which is produced by the Fenton reaction22–25) (Chart 1).
For the nitroxides resistant to AsA, the methyl radicals produced by the Fenton reaction were used. Rate constant, k was determined in the same way to that of AsA. To determine the concentration and conditions of the Fenton’s reagent to be used in the reaction with the nitroxides, a pre-examination using 20-, 100-, and 200-fold excess of Fenton’s reagent with 20 µM nitroxide 5 was carried out. It was found that 200-fold of reagent gave the best decay curve. The structure of the methyl adduct product was confirmed (see Experimental, compound 12). The reaction of nitroxide with methyl radicals is irreversible, unlike the conversion of nitroxide by AsA into hydroxylamine. The reactions of the five ibuprofen–nitroxides with 200-fold excess of Fenton’s reagent were conducted (Fig. 3). Since the appropriate decay curves were showed, the rate constants were calculated for 2, 4, and 5. Nitroxides 1 and 3 reacted immediately. The remaining sterically shielded nitroxides (2, 4, 5), which had hardly reacted with AsA, reacted in the order 2, 4, and 5. There was not a large difference between the rate constants. Nitroxides 2 and 4 reacted within 30 min, however, in the most sterically shielded nitroxide 5, 25% had not been reduced after 30 min. It was surprising that 5 was resistant not only to AsA but also to the labile methyl radicals.
Experimental
SynthesisMaterials and ApparatusAll starting materials and reagents were purchased from Wako Pure Chemical Industries, Ltd., Osaka, Japan and Sigma-Aldrich, St. Louis, MO, U.S.A. Reactions were monitored by TLC on 0.25-mm silica gel F254 plates (E. Merck, Japan). UV radiation and a 7% ethanolic solution of phosphomolybdic acid with heating, were used for coloration. Flash column chromatography was performed on silica gel (silica-gel 60, 40–50 µm, Kanto Chemical Co., Inc., Tokyo, Japan) to separate and purify the reaction products. Melting points were determined using an ASONE micro-melting point apparatus and uncorrected values reported. NMR spectra were recorded on a JEOL ECX-500 spectrometer using Me4Si as the internal standard. The 1H-NMR spectra of the hybrid compounds were measured and assigned for the corresponding hydroxylamines, which were prepared by the reduction of nitroxide radicals under the presence of excess of hydrazobenzene (Wako Pure Chemical Industries, Ltd.). Mass spectra and high-resolution mass spectra (HR-MS) were obtained under electron spray ionization (ESI) conditions on a JEOL JMS-T100LP.
General ProceduresSynthesis of Nitroxides 6–11The nitroxide radicals used in the present study were synthesized by established methods, as shown in Supplementary materials (Chart S1). Nitroxides 6, 7, 8, 9, and 10 were synthesized via 6, 4, 1, 4, and 3 steps from tetramethyl-4-piperidone in overall yield of 52, 17, 85, 62, and 0.8%, respectively. Nitroxide 11 was synthesized via 4 steps from tetraethyl-4-hydroxypiperidine with an overall yield of 6.05%. The synthetic yield of 10 was very low, 0.8%, because of the change of gem-methyl group to a gem-cyclohexyl group and the subsequent oxidation to nitroxide radical had a low unimprovable yield of 16 and 5.5%, respectively.
Synthesis of (2,2,6,6-Tetramethylpiperidin-1-oxyl)-4-yl (±)-2-(p-Isobutylphenyl)propionate (1)To a solution of ibuprofen (216 mg, 1.05 equiv.) and 3-hydroxymethyl-2,2,5,5-tetramethylpyperidin-1-oxyl (172 mg) in dry CH2Cl2 (6 mL), DCC (229 mg, 1.11 equiv.) and DMAP (13.5 mg, 0.111 equiv.) were added and the mixture was stirred at room temperature under an Ar atmosphere for 18 h. To the reaction mixture ethyl ether (20 mL) was added and the resulting precipitates were filtered through a celite pad. The filtrate was evaporated in vacuo and then purified by silica-gel column chromatography (n-hexane/ethyl acetate=3 : 1) to give 1 (360 mg, 100%) as red crystals.
The other ibuprofen nitroxides 2–5 were synthesized in a similar way to that of 1 in quantitative yields.
(2,2,6,6-Tetramethylpiperidin-1-oxyl)-4-yl (±)-2-(p-Isobutylphenyl)propionate (1)Red crystals, mp=48°C. ESI-MS (m/z) 362 (M+2H)+. 1H-NMR (CDCl3) δ (ibuprofen moiety) 0.89 (6H, d, J=6.8 Hz, CH3×2), 1.47 (3H, d, J=7.5 Hz, 2-CH3), 1.84 (1H, m, H8), 2.44 and 2.45 (each 1H, s, CH2), 3.64 (1H, q, J=7.5 Hz, H2), 7.08 and 7.18 (each 2H, d, J=8.2 Hz, ArH ×4), (piperidine moiety) 1.13, 1.15, 1.17, 1.18 (each 3H, s, –CH3×4), 1.45 and 1.54 (each 1H, t, J=11.6 Hz, CH2), 1.78 and 1.88 (each 1H, dt, J=0.4, 11.6 Hz, CH2), 5.02 (1H, m, >CH–). HR-MS (ESI+) m/z: [M + 2H]+, calcd for C22H36NO3 362.26952, found 362.26998.
(2,2,5,5-Tetramethyl-3-pyrroline-1-oxyl)-3-yl-methyl (±)-2-(p-Isobutylphenyl)propionate (2)Pale-yellow crystals, mp=45°C. ESI-MS (m/z) 381 (M+Na)+, 358 (M)+. 1H-NMR (CDCl3) δ (ibuprofen moiety) 0.89 (6H, d, J=6.7 Hz, CH3×2), 1.51 (3H, d, J=7.5 Hz, 2-CH3), 1.83 (1H, m, H8), 2.43 and 2.45 (each 1H, s, CH2), 3.72 (1H, q, J=7.5 Hz, H2), 7.09 and 7.21 (each 2H, d, J=8.3 Hz, ArH ×4), (piperidine moiety) 1.11, 1.12, 1.14, 1.15 (each 3H, s, –CH3×4), 4.52 and 4.61 (each 1H, d, J=14.1 Hz, CH2), 5.33 (1H, s, olefin H). HR-MS (ESI+) calcd for C22H32NO3 358.23822, found 358.23819.
(7-Aza-dispiro[5.1.5.3]hexadec-7-yl-oxyl)-15-yl (±)-2-(p-Isobutylphenyl)propionate (3)Pale-red crystals, mp=102°C. ESI-MS (m/z) 463 (M+Na)+, 442 (M+2H)+. 1H-NMR (CDCl3) δ (ibuprofen moiety) 0.88 (6H, d, J=6.8 Hz, CH3×2), 1.49 (3H, d, J=6.8 Hz, CH3), 1.84 (1H, m, H8), 2.43 and 2.45 (each 1H, s, CH2), 3.65 (1H, q, J=6.8 Hz, H2), 7.09 and 7.19 (each 2H, d, J=8.6 Hz, ArH ×4), (piperidine moiety) 1.00–1.45 (11H, m, cyclohexyl H), 1.51–1.68 (9H, m, cyclohexyl H), 2.24 and 2.35 (each 2H, br dt, J=12.1 Hz, CH2×2), 4.19 (1H, m, >CH–). HR-MS (ESI+) calcd for C28H42NNaO3 463.30624, found 463.30686.
(2,2,6,6-Tetraethylpiperidin-1-oxyl)-4-yl (±)-2-(p-Isobutylphenyl)propionate (4)Red powder. ESI-MS (m/z) 439 (M+Na)+. 1H-NMR (CDCl3) δ (ibuprofen moiety) 0.88 (6H, d, J=6.8 Hz, CH3×2), 1.47 (3H, d, J=6.8 Hz, 2-CH3), 1.28 (1H, m, H8), 2.42 and 2.44 (each 1H, s, CH2), 3.64 (1H, q, J=6.8 Hz, H2), 6.81 and 7.18 (each 2H, d, J=8.4 Hz, ArH ×4), (piperidine moiety) 0.78, 0.81, 0.82, 0.85 (each 3H, t, J=7.5 Hz, –CH2CH3×4), 1.24–1.30 (2H, –CH2CH3×1), 1.60–1.70 (4H, m, –CH2CH3×2), 1.60–1.70 (4H, m, –CH2CH3×2), 1.86 (2H, –CH2CH3×1), 1.43 and 1.97 (each 2H, t, J=9.1 Hz, CH2×2), 4.97 (1H, m, >CH–). HR-MS (ESI+) calcd for C26H42NNaO3 439.30624, found 439.30413.
(2,2,5,5-Tetraethyl-3-pyrroline-1-oxyl)-3-ylmethyl (±)-2-(p-Isobutylphenyl)propionate (5)Yellow powder. ESI-MS (m/z) 437 (M+Na)+. 1H-NMR (CDCl3) δ (ibuprofen moiety) 0.76 and 0.82 (each 3H, d, J=6.8 Hz, 8-CH3×2), 1.50 (3H, d, J=6.8 Hz, 2-CH3), 1.82 (1H, m, H8), 2.41 and 2.43 (each 1H, s, CH2), 3.72 (1H, q, J=6.8 Hz, H2), 6.81 and 7.19 (each 2H, d, J=6.8 Hz, ArH×4), (piperidine moiety) 0.87 and 0.89 (each 6H, br s, –CH2CH3×4), 1.40–1.62 (8H, m, –CH2CH3×4), 4.43 and 4.58 (each 1H, d, J=14.4 Hz, CH2), 5.24 (1H, s, olefin H). HR-MS (ESI+) calcd for C26H40NNaO3 437.29059, found 437.28977.
(1-Methoxy-2,2,5,5-tetramethyl-3-pyrrolin)-3-ylmethyl (±)-2-(p-Isobutylphenyl)propionate (12)To a solution of 2 (20 mg, 0.056 mmol) in DMSO (5 mL), 30% H2O2 aqueous solution (265 µL, 2.8 mmol) was added, with stirring. FeSO4·7H2O (770 mg, 2.8 mmol) was added by portions. After stirring for 1h, 50 mL of water was added to the reaction mixture and extracted with ethyl ether three times. The combined organic layer was washed with brine and dried over anhydrous Na2SO4. After removing the organic layer in vacuo, the residue was purified by silica-gel column chromatography (n-hexane/ether=10 : 1) to give 12 (17 mg, 82%) as a colorless powder.
Data of 12Colorless powder. ESI-MS (m/z) 396 (M+Na)+, 374 (M+H)+. 1H-NMR (CDCl3) δ (ibuprofen moiety) 0.89 (6H, d, J=6.8 Hz, CH3 ×2), 1.50 (3H, d, J=7.6 Hz, CH3), 1.84 (1H, m, H8), 2.43 and 2.45 (each 1H, s, CH2), 3.71 (1H, q, J=7.6 Hz, H2), 7.09 and 7.19 (each 2H, d, J=8.4 Hz, ArH ×4), (pyrroline moiety) 1.131 and 1.137 (each br s, CH3×2), 1.162 (6H, br s, CH3×2), 4.48 and 4.58 (each 1H, dd, J=1.5, 13.6 Hz, CH2), 5.25 (3H, s, OMe). 13C-NMR (CDCl3) δ (ibuprofen moiety) 18.39 (C9), 22.47 (C10), 30.31 (C8), 45.1 (C7), 45.3 (C2), 127.3 (C5), 129.4 (C4), 137.6 (3), 140.7 (C6), 174.4 (C1), (pyrroline moiety) 60.7 (CH2), 67.5, 69.7, 132.2, 139.1. HR-MS (ESI+) calcd for C23H36NNaO3 396.25146, found 396.25284.
AnalysisApparatusEPR spectra were obtained on a JEOL JES-FR30 EPR spectrometer. Samples were drawn into quartz capillaries, the bottoms of the capillaries were sealed and the capillaries were placed in standard 2-mm-i.d. quartz EPR tubes. The EPR spectrometer settings were as follows: microwave power, 4.0 mW; frequency, 9.5 GHz; and modulation amplitude, 1.25 G. Quick mixing of sample was conducted by VOLTEX®.
ReagentsThe reagents used in this study were commercial products: L-ascorbic acid (Wako, Japan), and dimethyl sulfoxide (DMSO; Wako, Japan), 30% H2O2 aqueous solution (Wako, Japan), and FeSO4·7H2O (Wako, Japan). DMSO was deoxygenated by microwave under reduced pressure and used.
EPR Intensity Measurement of Time-Dependent Reduction of Six Nitroxides with 200-Fold AsA in DMSOEach sample 1–6 and ascorbic acid were dissolved in DMSO to prepare 2 mM stock solutions. Twenty microliters of 2 mM sample and 1.98 mL of ascorbic acid in DMSO were mixed by VOLTEX® and an EPR measurement taken after 2 min. Subsequent measurements were taken at an interval of 1 min up to 10 min, and then the interval was changed to 2 min until 30 min had passed.
The conditions of the EPR measurement were: field, 336.5 mT; power, 4.0 mW; gain, 10.0; sweep width, 5.0 mT; modulation width, 0.1 mT; sweep time, 0.5 min; time constant, 0.1 s; date points, 0; accumulation, 1; accumulation method, no; and frequency, 9.2 GHz.
EPR intensity was calculated by following equation:
Twenty microliters of 2 mM nitroxide was diluted with 1.98 mL of DMSO to prepare 20 µM sample solution. EPR intensity of 20 µM sample solution was measured and the relative value of the EPR intensity was determined to be 1.0.
EPR Intensity Measurement of Time-Dependent Reduction of Five Nitroxides with Methyl Radicals in DMSOMethyl radical was prepared by the Fenton reaction. Thirty percent H2O2 aqueous solution was diluted with DMSO to prepare 4.04 mM H2O2 DMSO solution. Twenty microliters of each 2 mM sample DMSO solution and 1.98 mL of 4.04 mM H2O2 DMSO solution were mixed by VOLTEX® and the resulting mixture was used in the EPR measurement at 0 min. To this mixture, 2.2 mg of FeSO4·7H2O (8 µmol) was added and the mixture was mixed by VOLTEX® for 10 s. An EPR measurement was taken on the resulting mixture.
The EPR measurement and the determination of EPR intensity were conducted in the similar way to the above described.