Abstract
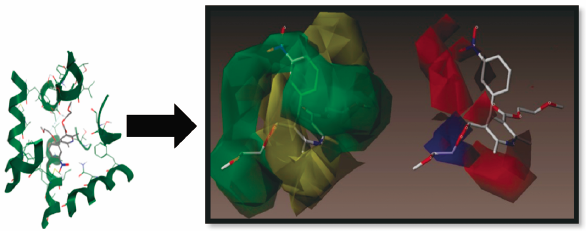
Resurgence to target L-type voltage-dependent calcium channels has been applied by the synthesis of two series of nifedipine analogues where the ortho- or a meta-nitrophenyl ring is retained. A pre-synthetic molecular docking study with a receptor model followed by molecular alignment has been performed on 47 compounds to predict the most active member. The IC50 values revealed that some of the compounds are similar to or more active than nifedipine. Substitution of groups at the 3- and 5-positions of the dihydropyridine (DHP) ring gave 3k, which is more active than nifedipine. Our valid three-dimensional quantitative structure–activity relationship (3D-QSAR) model prefigures the influence of lipophilicity, bulkiness and chelating effects of the C3 and C5 substituents. Bulky groups interfere with ring-to-ring hydrophobic interaction with tyrosine (Tyr)4311 and limit the efficiency of increasing the length of the hydrocarbon chain of esters at the 3- and 5-positions of the DHP ring as an approach to increasing the activity. The presence of a chelating substituent on the phenyl ring at the 4-position of the DHP ring ensures strong binding to the receptor and hence stabilization of the closed-channel conformation. The validation of 3D-QSAR model indicated its proficiency in predicting activity of newly compounds belonging to the same chemical class.