Experimental
GeneralSilica gel 60 (spherical and neutral; 100–210 µm, 37560-79) from Kanto Chemical Co. (Japan) was used for column chromatography. Preparative TLC was performed with Merck Silica Gel 60 F254 plates (0.5 mm thickness, No. 5744). Melting points were measured on a METTLER TOLEDO mp 70°C and were uncorrected. 1H-NMR spectra were measured at 400 MHz on a VARIAN 400-MR spectrometer or at 500 MHz on a VARIAN 500-MR spectrometer, and 13C-NMR spectra were measured at 125 MHz on a VARIAN 500-MR spectrometer. IR spectra were measured as attenuated total reflectance (ATR) on a Jasco Fourier transform (FT)/IR-4700 FT-IR spectrometer. High resolution (HR) MS were measured on Jeol JMS-T100LP AccuTOF. Optical rotation values were recorded on a Jasco P-1010 polarimeter.
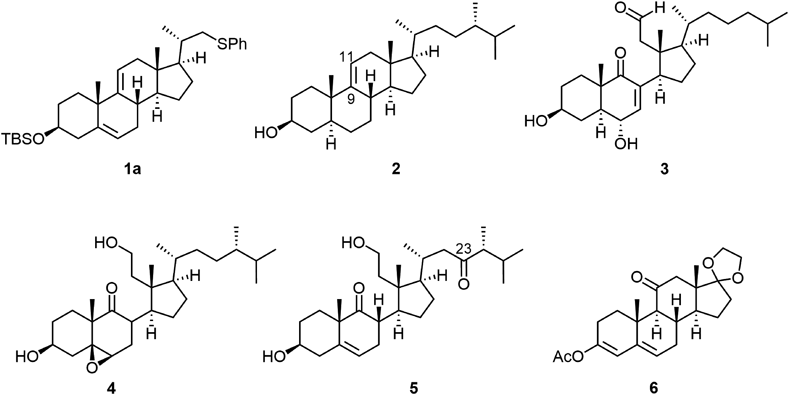
17-Ethylenedioxy-3-acetoxyandrosta-3,5-dien-11-one, 6According to the reported procedure,6) a solution of adrenosterone (2.44 g, 8.1 mmol, Tokyo Chemical Industry A1397) in acetic anhydride (20 mL) and acetyl chloride (1.4 mL) was stirred for 5 h at 100°C. After cooling, the white solid was recovered with filtration and washed with ice-cooled diethyl ether to give pure 3-acetoxyandrosta-3,5-diene-11,17-dione (2.12 g). In addition, the combined filtrate and washings were concentrated in vacuo and the residue was recrystallized from AcOEt to furnish an additional amount of the dienyl acetate (0.151 g) as a white solid. The total yield was 82%. Melting point (mp) 172.6–180.3°C (decomposed) (lit.,5) mp 197–198°C, lit.,6) mp 180–185°C). 1H-NMR (500 MHz, CDCl3) δ: 0.88 (s, 3H), 1.21 (s, 3H), 1.25 (ddd, J=5.6, 12.5, 12.5 Hz, 1H), 1.70 (dddd, J=9.5, 9.5, 12.5, 12.5 Hz, 1H), 1.91–1.97 (m, 2H), 2.00–2.06 (m, 1H), 2.09–2.22 (m, 3H), 2.14 (s, 3H), 2.28 (dd, J=9.0, 19.3 Hz, 1H), 2.34 (d, J=13.5 Hz, 1H), 2.44 (ddd, J=5.2, 5.2, 18.4 Hz, 1H), 2.48–2.60 (m, 3H), 2.67 (ddd, J=1.4, 5.6, 12.7 Hz, 1H), 5.39 (dd, J=2.7, 4.9 Hz, 1H), 5.69 (d, J=2.2 Hz, 1H). Its NMR spectrum was identical with that reported previously.5,6) This was employed for the next step without further purification.
To a solution of the above-mentioned dienyl acetate (2.62 g) in CH2Cl2 (25 mL) was added CH(OEt)3 (12.7 mL, 76.4 mmol), ethylene glycol (8.5 mL, 152 mmol) and p-toluenesulfonic acid (0.301 g, 3.7 mmol). The mixture was stirred for 2 h at room temperature. The reaction mixture was poured into saturated aqueous NaHCO3 and extracted with AcOEt. The extract was washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (60.0 g, hexane/AcOEt=30/1 to 7/1) to give 10 (2.63 g, 89%) as a white solid. An analytical sample was obtained by recrystallization from MeOH/acetone. Mp 133.3–134.5°C (decomposed) (lit.,5) mp 141.5–143.5°C). 1H-NMR (500 MHz, CDCl3) δ: 0.84 (s, 3H), 1.19 (s, 3H), 1.23 (ddd, J=5.6, 12.5, 12.5 Hz, 1H), 1.39 (dddd, J=6.1, 11.9, 11.9, 11.9 Hz, 1H), 1.80–1.87 (m, 1H), 1.91–2.14 (m, 8H), 2.13 (s, 3H), 2.31 (ddd, J=4.7, 18.3, 18.3 Hz, 1H), 2.47–2.54 (m, 1H), 2.63–2.68 (m, 2H), 3.83 (m, 2H), 3.93 (m, 2H), 5.36 (dd, J=2.7, 4.9 Hz, 1H), 5.67 (d, J=1.9 Hz, 1H). Its NMR spectrum was identical with that reported previously.5)
3β,11β-Dihydroxyandrost-5-en-17-one, 7aTo a suspension of NaBH4 (331 mg, 8.8 mmol) and CaCl2 (559 mg, 5.0 mmol) in EtOH (17.0 mL) was added a solution of 10 (582 mg) in CH2Cl2 (4.5 mL) dropwise at −15°C. After stirring for 12 h at −15°C, the mixture was warmed to room temperature and stirred for additional 13 h. To the mixture was added aqueous HCl (2 mol/L, 4.0 mL) and the mixture was stirred for further 2 h. To the mixture, AcOEt was added and the mixture was neutralized with saturated aqueous NaHCO3. After separation of the organic phase, the aqueous phase was extracted with AcOEt and the combined extract was washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (10.0 g, CHCl3/AcOEt=20/1 to 1/1) to give 11 (478 mg, quant.) as a white solid. An analytical sample was obtained by recrystallization from hexane/AcOEt. Mp 189.3–190.3°C (lit.,7) mp 190–192°C). 1H-NMR (500 MHz, CDCl3) δ: 1.06–1.09 (m, 2H), 1.14 (s, 3H), 1.20–1.30 (m, 2H), 1.29 (s, 3H), 1.46–1.61 (m, 3H), 1.64 (dddd, J=8.8, 8.8, 12.5, 12.5 Hz, 1H), 1.70–1.77 (m, 1H), 1.86–1.90 (m, 1H), 1.96–2.17 (m, 5H), 2.26–2.33 (m, 3H), 2.51 (dd, J=8.6, 18.8 Hz, 1H), 3.50–3.57 (m, 1H), 4.48–4.50 (m, 1H), 5.26–5.29 (m, 1H).
3β-tert-Butyldimethylsilyloxy-11β-hydroxyandrost-5-en-17-one, 7bTo a solution of 11 (742 mg, 2.4 mmol) and imidazole (376 mg, 5.5 mmol) in N,N-dimethylformamide (DMF) (10 mL) was added TBSCl (550 mg, 3.7 mmol). The mixture was stirred for 30 min at room temperature and then quenched with saturated aqueous NH4Cl, and the organic materials were extracted with AcOEt. The combined extract was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (20.0 g, hexane/AcOEt=30/1 to 8/1) to give 7b (976 mg, 96%) as a white solid. An analytical sample was obtained by recrystallization from hexane/AcOEt. Mp 190.6–191.6°C. 1H-NMR (500 MHz, CDCl3) δ: 0.06 (s, 6H), 0.89 (s, 9H), 1.05–1.08 (m, 2H), 1.13 (s, 3H), 1.17–1.28 (m, 2H), 1.28 (s, 3H), 1.48–1.79 (m, 5H), 1.94–2.37 (m, 8H), 2.50 (dd, J=8.8, 19.3 Hz, 1H), 3.46–3.52 (m, 1H), 4.47–4.50 (m, 1H), 5.24–5.25 (m, 1H). 13C-NMR (125 MHz, CDCl3) δ: −4.6, 15.7, 18.2, 21.7, 22.6, 25.9, 27.9, 31.1, 31.8, 35.3, 36.9, 36.9, 41.0, 41.9, 46.7, 53.7, 54.2, 68.2, 21.9, 119.5, 142.5, 219.6. IR cm−1: 3506, 2955, 2932, 2911, 2896, 2852, 1732, 1249, 1086, 1022, 889, 872, 832, 777, 514. HR-MS (electrospray ionization (ESI+)) Calcd for C25H42Na03Si [M+Na]+ 441.2801. Found 441.2777. [α]D25 −7.3 (c=0.96, CHCl3).
3β-tert-Butyldimethylsilyloxyandrosta-5,9(11)-dien-17-one, 8To a solution of 7b (471 mg, 1.1 mmol) in pyridine (11 mL) was added SOCl2 (130 µL, 1.8 mmol) at 0°C. The mixture was stirred for 20 min at the same temperature and then quenched with saturated aqueous NH4Cl. Organic materials were extracted several times with AcOEt. The combined extract was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (10.0 g, hexane/AcOEt=100/1 to 50/1) to give 8 (413 mg, 92%) as a white solid. An analytical sample was obtained by recrystallization from MeOH/acetone. Mp 154.9–156.3°C. 1H-NMR (500 MHz, CDCl3) δ: 0.06 (s, 6H), 0.87 (s, 3H), 0.89 (s, 9H), 1.20 (s, 3H), 1.36 (ddd, J=3.9, 13.1, 13.1 Hz, 1H), 1.49–1.86 (m, 5H), 1.95 (ddd, J=3.7, 13.0, 13.0 Hz, 1H), 2.02–2.40 (m, 8H), 2.49 (dd, J=9.0, 18.0 Hz, 1H), 3.45–3.52 (m, 1H), 5.43–5.44 (m, 1H), 5.53 (d, J=5.9 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ: −4.6, 13.7, 18.3, 22.8, 25.9, 27.7, 31.5, 32.2, 33.5, 34.0, 35.0, 36.5, 38.7, 42.7, 46.3, 49.0, 72.6, 116.0, 120.1, 140.1, 146.7, 221.8. IR cm−1: 2927, 2905, 2897, 2856, 2842, 1741, 1470, 1445, 1436, 1381, 1370, 1362, 1251, 1221, 1079, 1061, 1032, 1007, 992, 973, 964, 937, 884, 870, 852, 835, 820, 806, 772, 732, 668, 638. HR-MS (ESI+) Calcd for C25H40NaO2Si [M+Na]+ 423.2695. Found 423.2668. [α]D24 +239.4 (c=1.02, CHCl3).
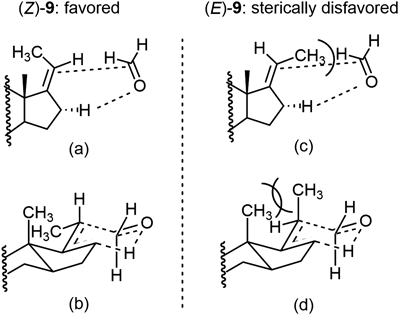
3β-tert-Butyldimethylsilyloxypregna-5,9(11),17(20)-triene, 9To a solution of ethyltriphenylphosphonium bromide (432 mg, 1.2 mmol) in tetrahydrofuran (THF) (1 mL) was added a solution of KHMDS in toluene (0.5 mol/L, 2.3 mL, 1.2 mmol) at 0°C. The mixture was stirred for 2 h at 0°C, and then a solution of 8 (103 mg, 0.26 mmol) in THF (2 mL) was added to that. The mixture was stirred under reflux for 18 h. After cooling to room temperature, the reaction was quenched with water and the organic materials were extracted with AcOEt. The extract was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (7.5 g, hexane/AcOEt=200/1 to 50/1) to give 9 (106 mg, quant.) as a white solid. 1H-NMR (500 MHz, CDCl3) δ: 0.07 (s, 6H), 0.75 [s, 3H, C18 of (E)-9], 0.86 [s, 3H, C18 of (Z)-9], 0.89 (s, 9H), 1.19 (s, 3H), 1.23–1.40 (m, 3H), 1.56 [ddd, J=1.5, 1.5, 6.6 Hz, 3H, 21-C of (E)-9], 1.61–1.85 (m, 4 H), 1.66 [ddd, J=2.2, 2.2, 7.3 Hz, 3H, C21 of (Z)-9], 1.95 (ddd, J=3.4, 3.4, 13.2 Hz, 1H), 2.04–2.49 (m, 8H), 3.44–3.52 (m, 1H), 5.09 [qdd, J=6.6, 3.5, 3.5 Hz, 1H, C20 of (E)-7], 5.21 [qdd, J=7.1, 2.0, 2.0 Hz, 1H, C20 of (Z)-9], 5.42–5.43 (m, 1H), 5.48–5.50 [m, 1H, C11 of (Z)-9], 5.53–5.54 [m, 1H, C11 of (E)-9]. Judged from the above-mentioned 1H-NMR spectrum, the ratio between (Z)- and (E)-9 was revealed to be 5 : 1.
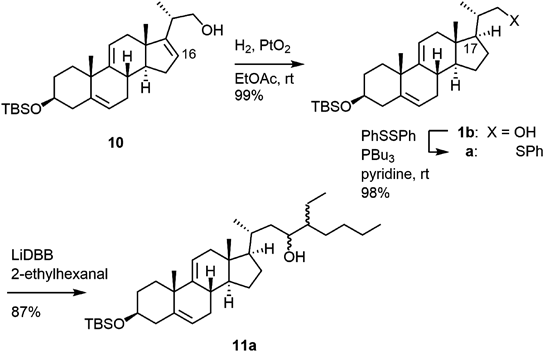
3β-tert-Butyldimethylsilyloxy-23,24-bisnorchola-5,9(11),16-trien-22-ol, 10To a mixture of 9 [71 mg, 0.18 mmol as a mixture of (Z) : (E)=5 : 1] and paraformaldehyde (35 mg, 1.2 mmol) in CH2Cl2 (2.5 mL) was added a solution of BF3·Et2O in CH2Cl2 (0.8 mol/L, 0.03 mmol) at room temperature. The mixture was stirred for 30 min at the same temperature. Then, the reaction was quenched with saturated aqueous NaHCO3 and the organic materials were extracted several times with AcOEt. The combined extract was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (1.6 g, hexane/AcOEt=50/1 to 2/1) to give 10 as a white solid (48 mg, 63%) also unreacted 9 was recovered (15%). An analytical sample of 10 was obtained by recrystallization from hexane. Mp 132.3–133.5°C. 1H-NMR (500 MHz, CDCl3) δ: 0.06 (s, 3H), 0.78 (s, 3H), 0.89 (s, 9H) 1.07 (d, J=7.1 Hz, 3H), 1.20 (s, 3H), 1.36 (ddd, J=3.7, 13.7, 13.7 Hz, 1H), 1.44 (dd, J=4.9, 7.3 Hz, 1H), 1.61–1.71 (m, 2H), 1.82–1.84 (m, 1H), 1.91–2.05 (m, 3H), 2.19–2.44 (m, 7H), 3.46–3.52 (m, 1H), 3.54–3.64 (m, 2H), 5.42–5.43 (m, 1H), 5.47–5.51 (m, 2H). 13C-NMR (125 MHz, CDCl3) δ: −4.6, 15.6, 18.3, 18.3, 25.9, 27.6, 31.9, 32.0, 32.3, 32.4, 35.0, 35.4, 37.1, 38.6, 42.7, 46.1, 54.2, 66.8, 72.7, 116.4, 120.5, 123.3, 139.9, 147.6, 155.8. IR cm−1: 3274, 3033, 3018, 2956, 2929, 1891, 2857, 1471, 1457, 1446, 1437, 1364, 1252, 1219, 1092, 1081, 1068, 1034, 1017, 1005, 964, 889, 871, 835, 818, 809, 769, 685, 668, 647, 627. HR-MS (ESI+) Calcd for C28H46NaO2Si [M+Na]+ 465.3165. Found 465.3160. [α]D26 +4.5 (c=1.15, CHCl3). The recovered 9 showed exclusive (E)-configuration. Two dimensional (2D)- nuclear Overhauser effect spectroscopy (NOESY) showed the correlation between the protons at C12 and C20. The purity was confirmed by 1H-NMR mentioned in the previous section.
3β-tert-Butyldimethylsilyloxy-23,24-bisnorchola-5,9(11)-dien-22-ol, 1bA mixture of 10 (48 mg, 0.11 mmol) and PtO2 (8 mg, 0.04 mmol) in AcOEt (7 mL) was stirred for 8 h under H2 atmosphere at room temperature. After the removal of insoluble materials, the combined filtrate and washings were concentrated in vacuo. The residue was purified by preparative TLC to give 1b (48 mg, 99%) as a white solid. An analytical sample was obtained by recrystallization from hexane. Mp 134.3–135.3°C. 1H-NMR (500 MHz, CDCl3) δ: 0.06 (s, 6H), 0.66 (s, 3H), 0.89 (s, 9H), 1.05 (d, J=6.6 Hz, 3H), 1.16–1.42 (m, 6H), 1.18 (s, 3H), 1.56–1.69 (m, 3H), 1.74–1.90 (m, 3H), 1.93 (ddd, J=3.4, 13.4, 13.4 Hz, 1H), 2.02–2.29 (m, 6H), 3.37–3.42 (m, 1H), 3.45–3.51 (m, 1H), 3.64–3.68 (m, 1H), 5.40–5.44 (m, 2H). 13C-NMR (125 MHz, CDCl3) δ: −4.6, 11.4, 16.4, 18.3, 25.4, 25.9, 27.6, 28.0, 32.3, 32.4, 34.3, 35.0, 38.3, 38.6, 41.3, 41.8, 42.7, 52.4, 53.4, 68.0, 72.7, 116.9, 120.7, 139.9, 146.1. IR cm−1: 3307, 2955, 2928, 2898, 2881, 2855, 1470, 1458, 1437, 1382, 1254, 1220, 1080, 1038, 1027, 1005, 987, 885, 871, 838, 819, 806, 775, 769, 685, 673. HR-MS (ESI+) Calcd for C28H48NaO2Si [M+Na]+ 467.3321. Found 467.3321. [α]D26 −14.7 (c=0.98, CHCl3).
3β-tert-Butyldimethylsilyloxy-22-phenylthio-23,24-bisnorchola-5,9(11)-diene, 1aTo a solution of 1b (758 mg, 1.7 mmol) and diphenyl disulfide (1.125 g, 5.2 mmol) in pyridene (34 mL) was added tri-n-butylphosphine (2.7 mL, 10.9 mmol) under Ar atmosphere. The mixture was stirred for 19 h at room temperature and then quenched with saturated aqueous NH4Cl. The organic materials were extracted several times with AcOEt. The combined extract was washed with aqueous NaOH (2 mol/L) twice and brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (40.0 g, hexane/AcOEt=100/1 to 70/1) to give 1a (898 mg, 98%) as a white solid. An analytical sample was obtained by recrystallization from MeOH/acetone. Mp 134.3–135.3°C. 1H-NMR (500 MHz, CDCl3) δ: 0.06 (s, 6H), 0.63 (s, 3H), 0.89 (s, 9H), 1.12 (d, J=6.6 Hz, 3H), 1.15–1.40 (m, 5H), 1.17 (s, 3H), 1.60–1.69 (m, 2H), 1.74–1.83 (m, 3H), 1.90–2.29 (m, 8H), 2.66 (dd, J=8.8, 12.2 Hz, 1H), 3.16 (dd, J=3.0, 12.2 Hz, 1H), 3.44–3.51 (m, 1H), 5.40–5.43 (m, 2H), 7.05–7.33 (m, 5H). 13C-NMR (125 MHz, CDCl3) δ: −4.6, 11.3, 18.3, 18.6, 25.3, 25.9, 27.6, 28.4, 32.3, 32.4, 34.3, 35.0, 36.3, 38.3, 41.1, 41.5, 41.7, 42.7, 53.5, 55.5, 72.7, 103.3, 116.8, 120.7, 125.5, 128.8, 137.8, 139.9, 146.1. IR cm−1: 3734, 3648, 2955, 2935, 2925, 2901, 2892, 2879, 2854, 1541, 1458, 1437, 1220, 1093, 1083, 774, 768, 743, 688, 673. Elemental analysis: Calcd for C34H52OSSi: C, 76.07; H, 9.77; S, 5.96. Found: C. 76.22; H, 10.04; S, 6.21. [α]D23 +20.3 (c=1.05, CHCl3).
24-Butyl-3β-tert-butyldimethylsilyloxybishomochola-5,9(11)-dien-23-ol, 11aA solution of LiDBB in THF (0.4 mol/L) was prepared, according to the reported procedure.20) To a solution of 1a (211 mg, 0.39 mmol) in THF (5.5 mL) was add a solution of LiDBB in THF (0.4 mol/L, 1.3 mmol) at −78°C dropwise over 20 min. The low reaction temperature was carefully controlled by elaborating the way of addition. The solution of pre-cooled (0°C) of LiDBB in THF was slowly introduced via the side wall of two-necked flask, which was deeply dipped in the cold bath. The mixture was stirred for 10 min and 2-ethylhexanal (0.6 mL, 3.8 mmol) was added to that at the same temperature dropwise over 10 min. After stirring 24 h, the reaction temperature was raised to room temperature and the mixture was stirred for additional 1 h. The reaction was quenched with saturated aqueous NH4Cl, and the organic materials were extracted several times with AcOEt. The combined extract was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (20.0 g, hexane/AcOEt=1/0 to 50/1) to give 11a (191 mg, 87%). 1H-NMR (CDCl3, 500 MHz) δ: 0.06 (s, 6H), 0.65 (s, 3H), 0.89–2.29 (m, 45H), 3.44–3.51 (m, 1H), 3.69–3.82 (m, 1H), 5.40–5.43 (m, 2H). The structure was further confirmed by acetylation to acetate 11b. Rf value of 11a (0.37, developed with a mixture of hexane/AcOEt=10/1) was changed to 0.57 for 11b. The signal δ: 3.69–3.82 in 11a was downfield-shifted to 5.04–5.09 in 11b. HR-MS (ESI+) Calcd for C38H66NaO3Si [M+Na]+ 621.4679. Found 621.4678.