Regular Articles
Palladium-Catalyzed External-CO-Free Carbonylation of Aryl Bromides Using 2,4,6-Trichlorophenyl Formate
2018 Volume 66 Issue 5 Pages 562-567

Details
2018 Volume 66 Issue 5 Pages 562-567
A practical Pd-catalyzed carbonylation of (hetero)aryl bromides using a crystalline carbon monoxide (CO) surrogate, 2,4,6-trichlorophenyl formate (TCPF), was developed. This reaction proceeds without the slow addition technique that was previously required and with a low catalyst loading (1 mol%). The utility of this Pd-catalyzed external-CO-free carbonylation using TCPF was demonstrated in the synthesis of a histone deacetylase inhibitor.
Aromatic carboxylic acid derivatives are important compounds that comprise many drugs, fine chemicals, and their synthetic intermediates. Aromatic halides constitute useful starting materials in the synthesis of aromatic carboxylic acid derivatives. There are mainly two synthetic approaches that can be used to obtain the carboxylic acid derivatives from aromatic halides: metal–halogen exchange using metals or organometallic reagents followed by treatment with carbon dioxide1–3) and derivatization of the resulting carboxylic acids, and Pd-catalyzed carbonylation in the presence of nucleophiles.4–7) The former method involves a strongly basic metalated intermediate that is incompatible with sensitive functional groups, while the latter proceeds smoothly under weakly basic conditions.
Because of the versatility of Pd-catalyzed carbonylation, it has been widely studied since the pioneering work of Heck and co-workers reported in 1974.8–10) However, because the reaction uses toxic and flammable carbon monoxide (CO) gas, special cautions are required, especially when the reaction is conducted on a laboratory scale. To solve this problem, many research groups have worked on the development of CO surrogates that generate CO in situ, and several safe and facile carbonylation reactions using CO surrogates have been reported.11–18)
Recently, we and Tsuji et al. independently reported Pd-catalyzed carbonylation reactions using a liquid CO surrogate, phenyl formate. It was found to react with weak bases (e.g., NEt3) at elevated temperatures to form CO and phenol, and could be employed in external-CO-free carbonylation reactions in situ.19–21) In the course of our investigations on more reactive CO surrogates, we also developed a highly reactive crystalline CO surrogate, 2,4,6-trichlorophenyl formate (1, TCPF).22) While it is highly air-stable and can be stored under ambient conditions, the generation of CO from 1 occurs rapidly (in ca. 30 min) in the presence of NEt3, even at room temperature (r.t.). The utilization of 1 enabled us to conduct the Pd-catalyzed aryloxycarbonylation of iodoarenes and alkenyl triflates at r.t. We also reported facile, scalable procedures to prepare TCPF from low-priced starting materials and their applications in Gram-scale Pd-catalyzed carbonylation reactions.23) Furthermore, the resulting aryloxycarbonylated products, trichlorophenyl esters, are electrophilic enough to be easily transformed with various nucleophiles to give other derivatives such as amides and thioesters in high yields.24,25) Therefore, carbonylation using 1 and subsequent transformations can be useful for synthesizing various carboxylic acid derivatives.22,26–28)
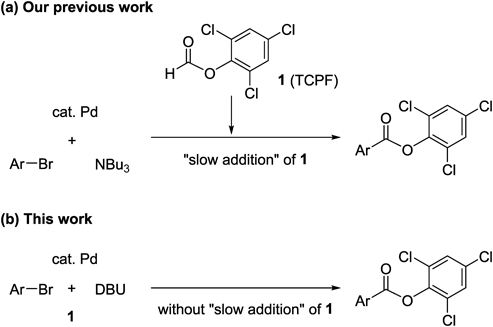
Despite the advantages of 1, the carbonylation of bromoarenes, which are considerably cheaper than iodoarenes and are readily available, requires high temperatures (100°C) and the slow addition of a solution of 1 (over 3 h) to the reaction mixture22) (Chart 1a). This procedure is not practical, since a syringe pump is required for the slow addition. Herein, we present a practical Pd-catalyzed external-CO-free carbonylation of bromoarenes using TCPF, without the use of a syringe pump (Chart 1b). In addition, this improved procedure can also be conducted with a smaller amount of the Pd catalyst.
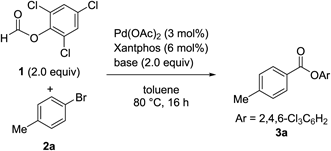
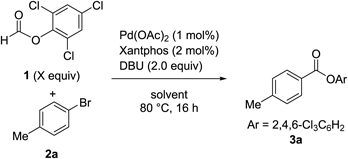
To identify the optimal conditions for the carbonylation of bromoarenes using TCPF without the use of a syringe pump, we examined the effects of various bases on the carbonylation of 4-bromotoluene (2a) using 2.0 eq of 1 and a catalytic amount of Pd(OAc)2 and Xantphos in toluene at 80°C (Table 1). The reactions were conducted in a test tube, and all materials were added before the tube was sealed with a screw cap and heated in an oil bath. Organic (entries 1–7) and inorganic bases (entries 9–16) were screened. NEt3 or NBu3, which were used in the previous work,22) resulted in relatively good yields, albeit not very high (entries 4 and 5). Interestingly, sterically bulky iPr2NEt was totally ineffective (entry 6). Among all the tested bases, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) proved to be optimal (entry 7), affording 3a in a high yield. Furthermore, with DBU as a base, the carbonylated product was obtained in 78% yield with 1 mol% of Pd(OAc)2 (entry 8).
 | ||
|---|---|---|
| Entry | Base | Yield (%) |
| 1 | NaOAc | Trace |
| 2 | Pyridine | 6 |
| 3 | DABCO | 26 |
| 4 | NEt3 | 79 |
| 5 | NBu3 | 83 |
| 6 | iPr2NEt | Trace |
| 7 | DBU | 93 |
| 8a) | DBU | 78 |
| 9 | K2HPO4 | Trace |
| 10 | K3PO4 | 19 |
| 11 | NaHCO3 | 13 |
| 12 | Na2CO3 | 58 |
| 13 | Li2CO3 | Trace |
| 14 | K2CO3 | Trace |
| 15 | Rb2CO3 | 48 |
| 16 | Cs2CO3 | 79 |
a) Pd(OAc)2 (1 mol%) and Xantphos (2 mol%) were used.
Next, we tested various solvents in the carbonylation reaction using 1.5 eq of 1 in the presence of 1 mol% of Pd(OAc)2, 2 mol% of Xantphos, and 2.0 eq of DBU (Table 2, entries 1–8). Less polar solvents appeared to be better suited for this reaction than polar solvents, and toluene was determined to be the optimal solvent. We observed too fast generation of CO during the addition of DBU when polar solvents were used, which might cause a CO leak through the hole made on a septum by piercing the syringe. We also examined the effect of the amount of 1 (entries 8–10), because 1 generates CO rapidly in the reaction with DBU in toluene at 80°C, therefore the amount of 1 affects the CO pressure inside the sealed reaction vessel. Since high CO pressure often retards the oxidative addition of bromoarenes to the Pd center due to the π-acidity of CO,29,30) the internal CO pressure would affect the Pd-catalyzed process. Two equivalents of 1 were optimal for the reaction (entry 9), and 1.5 or 3.0 eq of 1 gave slightly lower yields of the desired ester (entries 8 and 10). These results suggest that appropriate CO pressure is important for the reaction to proceed smoothly. Pd sources were also screened, and Pd(PhCN)2Cl2 was found to be a more effective catalyst than Pd(OAc)2, affording the desired product in >99% yield (entry 11).
 | |||
|---|---|---|---|
| Entry | Solvent | X (eq) | Yield (%) |
| 1 | CH3CN | 1.5 | 62 |
| 2 | DMF | 1.5 | 43 |
| 3 | NMP | 1.5 | 13 |
| 4 | DCE | 1.5 | 35 |
| 5 | PhCF3 | 1.5 | 67 |
| 6 | THF | 1.5 | 62 |
| 7 | DME | 1.5 | Trace |
| 8 | Toluene | 1.5 | 72 |
| 9 | Toluene | 2.0 | 78 |
| 10 | Toluene | 3.0 | 65 |
| 11a) | Toluene | 2.0 | >99 |
a) Pd(PhCN)2Cl2 was used instead of Pd(OAc)2.
With the optimal reaction conditions in hand, we examined the substrate scope of the reaction (Table 3). The reaction proceeded in 75–91% yields (3a–g) with substrates bearing a variety of para-substituents such as methoxy, chloro, ethoxycarbonyl, formyl, and cyano groups. In contrast, the substrate bearing a nitro group resulted in a modest yield of 3h (34%). meta-Substituted substrates bearing methyl and methoxy groups reacted with 2 to provide 3i and 3j in 83–91% yield. On the other hand, the carbonylation of ortho-substituted bromoarenes with methyl and chloro groups was sluggish, yielding the product in 24–52% yields (3k, l), probably due to the steric effects of the substituents. However, to our delight, carbonylation of the ortho cyano substrate gave the product in 84% yield (3m). While 1-bromonaphthalene gave the product in 59% yield (3n), the reaction of 2-bromonaphthalene gave the product in >99% yield (3o). Furthermore, the reactions of electron-deficient bromoheteroarenes, 3-bromopyridine and 5-bromopyrimidine, proceeded smoothly to give the product in 89% (3p) and 67% yields (3q), respectively. Electron-rich bromoheteroarenes such as 3- and 2-bromothiophene reacted with 1 to afford the corresponding esters in 88–89% yields (3r, s). The carbonylation reaction of phenyl triflate afforded the desired product in 79% yield. In addition, the carbonylation reaction was applied to the gram-scale synthesis of 3a, and 1.04 g (66% yield) of the desired product was obtained in a single run. The reaction with 0.5 mol% of the Pd catalyst gave 3a in 79% yield in 48 h.
 |
a) The reaction was conducted in 5 mmol scale. 1.04 g of the product was obtained. b) Pd(PhCN)2Cl2 (0.5 mol%) and Xantphos (1 mol%) were used. The reaction was performed for 48 h.
Finally, to demonstrate the utility of the reaction, we applied it to the synthesis of a histone deacetylase (HDAC) inhibitor (7) developed by Qvortrup and Nielsen (Chart 2) via solid-phase synthesis.31) Bromothiophene 4, which was prepared from (5-bromothiophen-2-yl)methanamine32) and 4-fluorobenzoyl chloride, smoothly reacted with 1 to afford trichlorophenyl ester 5 in 93% yield on a gram scale. Ester 5 was then condensed with (S)-O-benzyl-α-alaninehydroxamic acid trifluoroacetic acid (TFA) salt to give amide 6. Deprotection of the benzyl group with BCl3 gave 7. Thus, the carbonylation reaction using TCPF enabled the efficient solution-phase synthesis of HDAC inhibitor 7.

Reagents and conditions: (i) 4 (1.00 g, 3.18 mmol), 1 (2.0 eq), DBU (2.0 eq), Pd(PhCN)2Cl2 (1 mol%), Xantphos (2 mol%), toluene, 80°C, 24 h, 93% (1.35 g of 5 was obtained.); (ii) (S)-O-benzyl-α-alaninehydroxamic acid TFA salt, NEt3, DMAP (5 mol%), THF, 55°C, 44 h, 92%; (iii) BCl3, CH2Cl2, 0°C, 1 h, 49%.
In summary, we developed the Pd-catalyzed carbonylation of bromoarenes and an aryl triflate using TCPF, an easily accessible crystalline CO surrogate, without the previously required slow and crystalline technique. Moreover, the reaction could be conducted with a low catalyst loading (1 mol%) on a gram scale. The developed carbonylation reaction does not require a syringe pump, and can be conducted in a simple test tube or a flask. Therefore, this method provides a practical, safe, and easy route for the transformation of bromoarenes into trichlorophenyl esters with a wide viable substrate scope including heteroarenes. Furthermore, the synthesis of an HDAC inhibitor was realized in only a few short steps from a bromothiophene derivative, highlighting the potential utility of this reaction. Further investigations on other carbonylative reactions using this CO surrogate are underway and will be reported in due course.
All reactions were performed in oven-dried or flame-dried glassware under an argon atmosphere. Reactions were monitored by TLC on Merck silica gel 60 F254 plates visualized with a UV lamp at 254 nm. Column chromatography was performed on Merck silica gel 60, and preparative TLC was performed on Merck silica gel 60 F254 0.5 mm plates. NMR spectra were measured on a JEOL AL-400 NMR spectrometer (400 MHz for 1H spectra and 100 MHz for 13C spectra) or a JEOL ECX-500 NMR spectrometer (500 MHz for 1H spectra and 125 MHz for 13C spectra) and are quoted in ppm for measurement against a tetramethylsilane (TMS) or residual solvent peak as an internal standard. Infrared spectra were measured on a SHIMADZU IR Prestige-21 spectrometer (attenuated total reflection (ATR)). High-resolution (HR)-MS were measured on a Bruker MicrOTOF time-of-flight (TOF)-MS electrospray ionization (ESI) and a JEOL JMS-T100TD TOF-MS (DART). Melting points were measured using a YAZAWA MICRO MELTING POINT BY-1.
General Experimental Procedure for Carbonylation of Bromoarenes Using 2,4,6-Trichlorophenyl Formate (1)Pd(PhCN)2Cl2 (1.9 mg, 5.00 µmol, 1 mol%), Xantphos (1.9 mg, 10.0 µmol, 2 mol%), bromoarene (0.500 mmol), 123) (226 mg, 1.00 mmol, 2.0 eq), and toluene (1.0 mL) were added to a 10 mL test tube with a septum containing a magnetic stirring bar. The tube was evacuated and backfilled with Ar three times. DBU (150 µL, 1.00 mmol, 2.0 eq) was added to the mixture using a syringe through the septum. The tube was screw-capped and warmed to 80°C in an oil bath. The mixture was stirred for 24 h. After cooling to r.t., the mixture was quenched with aq. citric acid (10% (w/v)), diluted with CH2Cl2 and H2O, extracted with CH2Cl2 from aqueous layer, washed with H2O, aq. Na2CO3 (0.5 M), and brine, dried over Na2SO4, filtered, and concentrated. The obtained residue was purified by preparative TLC (SiO2, hexane–EtOAc 30 : 1) to afford the desired ester.
2,4,6-Trichlorophenyl 4-Methylbenzoate (3a)22)White solid; 158 mg, >99% yield.
2,4,6-Trichlorophenyl Benzoate (3b)22)White solid; 135 mg, 90% yield.
2,4,6-Trichlorophenyl 4-Methoxybenzoate (3c)22)White solid, 125 mg, 75% yield.
2,4,6-Trichlorophenyl 4-Chlorobenzoate (3d)22)White solid, 147 mg, 88% yield.
2,4,6-Trichlorophenyl 4-Ethoxycarbonylbenzoate (3e)22)Pale yellow solid, 174 mg, 93% yield.
2,4,6-Trichlorophenyl 4-Formylbenzoate (3f)22)Pale yellow solid; 143 mg, 85% yield.
2,4,6-Trichlorophenyl 4-Cyanobenzoate (3g)22)Pale yellow solid, 148 mg, 91% yield.
2,4,6-Trichlorophenyl 4-Nitrobenzoate (3h)White solid; 58.8 mg, 34% yield; mp 105.0–105.3°C; 1H-NMR (400 MHz, CDCl3) δ: 8.44–8.38 (m, 4H), 7.45 (s, 2H) ppm; 13C-NMR (125 MHz, CDCl3) δ: 161.3, 151.4, 142.8, 133.3, 132.8, 131.8, 129.6, 128.9, 124.0 ppm; IR (ATR) 1751, 1524, 1254, 1227, 1180, 1134, 1057, 1011, 980, 845, 802, 710, 667, 571, 498, 451 cm−1, Anal. Calcd for C13H6Cl3NO4 C, 45.06; H, 1.75; N, 4.04. Found: C, 44.84; H, 1.71; N, 4.10.
2,4,6-Trichlorophenyl 3-Methylbenzoate (3i)White solid; 132 mg, 83% yield; mp 115.3–116.1°C; 1H-NMR (400 MHz, CDCl3) δ: 8.05–8.03 (m, 2H), 7.49–7.40 (m, 4H), 2.45 (s, 3H) ppm; 13C-NMR (125 MHz, CDCl3) δ: 163.2, 143.4, 138.8, 135.2, 132.1, 131.1, 129.9, 128.8, 128.7, 127.9, 127.9, 21.4 ppm; IR (ATR) 1750, 1449, 1270, 1235, 1187, 1139, 1045, 862, 824, 802, 731, 562, 512, 507, 502 cm−1; HR-MS (DART) [M+H]+ Calcd for C14H10Cl3O2: 314.9746. Found 314.9753.
2,4,6-Trichlorophenyl 3-Methoxylbenzoate (3j)White solid; 151 mg, 91% yield; mp 60.3–61.2°C; 1H-NMR (400 MHz, CDCl3) δ: 7.85 (dt, J=7.6, 1.2 Hz, 1H), 7.72 (t, J=1.6 Hz, 1H), 7.45 (t, J=8.4 Hz, 1H), 7.42 (s, 2H), 7.22 (ddd, J=8.4, 2.8, 1.2 Hz, 1H), 3.89 (s, 3H) ppm; 13C-NMR (125 MHz, CDCl3) δ: 163.0, 159.9, 143.3, 132.2, 129.9, 129.2, 128.7, 123.1, 121.0, 114.8, 55.6 ppm; IR (ATR) 1740, 1273, 1234, 1204, 1177, 1138, 1026, 907, 856, 822, 802, 741, 563, 498, 451, 420 cm−1; HR-MS (DART) [M+H]+ Calcd for C14H10Cl3O3: 330.9696. Found 330.9691.
2,4,6-Trichlorophenyl 2-Methylbenzoate (3k)22)White solid; 38.1 mg, 24% yield.
2,4,6-Trichlorophenyl 2-Chlorobenzoate (3l)22)White solid; 86.9 mg, 52% yield.
2,4,6-Trichlorophenyl 2-Cyanobenzoate (3m)White solid; 137 mg, 84% yield; mp 156.9–158.3°C; 1H-NMR (400 MHz, CDCl3) δ: 8.43–8.41 (m, 1H), 7.94–7.92 (m, 1H), 7.82–7.80 (m, 2H), 7.44 (s, 2H) ppm; 13C-NMR (125 MHz, CDCl3) δ: 160.4, 142.6, 135.4, 134.1, 132.9, 132.7, 132.1, 129.9, 129.7, 128.8, 116.9, 114.0 ppm; IR (ATR) 1753, 1443, 1252, 1227, 1119, 1047, 1028, 853, 820, 789, 760, 727, 685, 658, 565, 552, 527, 509, 501 cm−1; HR-MS (DART) [M+H]+ Calcd for C14H7Cl3NO2: 325.9542. Found 325.9535.
2,4,6-Trichlorophenyl 1-Naphthoate (3n)22)White solid; 104 mg, 59% yield.
2,4,6-Trichlorophenyl 2-Naphthoate (3o)22)White solid; 156 mg, 89% yield.
2,4,6-Trichlorophenyl Nicotinate (3p)22)White solid; 135 mg, 89% yield.
2,4,6-Trichlorophenyl Pyrimidine-5-carboxylate (3q)22)White solid; 96.6 mg, 64% yield.
2,4,6-Trichlorophenyl Thiophene-3-carboxylate (3r)6)White solid; 140 mg, 89% yield.
2,4,6-Trichlorophenyl Thiophene-2-carboxylate (3s)White solid; 136 mg, 88% yield; mp 63.4–63.8°C; 1H-NMR (400 MHz, CDCl3) δ: 8.05 (dd, J=4.0, 0.8 Hz, 1H), 7.73 (dd, J=4.8, 0.8 Hz, 1H), 7.41 (s, 2H), 7.21 (dd, J=4.8, 4.0 Hz, 1H) ppm; 13C-NMR (125 MHz, CDCl3) δ: 158.3, 142.9, 135.9, 134.7, 132.3, 130.8, 130.1, 128.7, 128.4 ppm; IR (ATR) 1732, 1562, 1450, 1408, 1354, 1261, 1227, 1138, 1076, 1053, 999, 856, 802, 725, 698, 563, 478, 451, 440 cm−1; HR-MS (DART) [M+H]+ Calcd for C11H6Cl3O2S: 306.9154. Found 306.9159.
Synthesis of N-((5-Bromothiophen-2-yl)methyl)-4-fluorobenzamide (4)A solution of 4-fluorobenzoyl chloride (1.06 g, 6.69 mmol, 1.2 eq) in CH2Cl2 (2.5 mL) was added to a stirred solution of (5-bromothiophen-2-yl)methanamine32) (1.09 g, 5.65 mmol) and iPr2NEt (1.73 mL, 10.2 mmol, 1.8 eq) in CH2Cl2 (8.2 mL) in 50-mL round bottomed flask at 0°C over 30 min, and then the reaction was allowed to warm r.t. over 19.5 h. The mixture was diluted with CH2Cl2, washed with sat. aq. NaHCO3 (twice), dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude brown solid was purified by column chromatography (SiO2, hexane–EtOAc=4 : 1 to 1 : 1). Since the resulting brown solid was found to be contaminated with 4-fluorobenzoic acid, the solid was dissolved in EtOAc, washed with aq. Na2CO3 (0.5 M), brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to yield 4 as an off white solid (1.51 g, 4.81 mmol, 85%). mp 116.4–117.1°C; 1H-NMR (400 MHz, CDCl3) δ: 7.80–7.77 (m, 2H), 7.13–7.08 (m, 2H), 6.90 (d, J=3.2 Hz, 1H), 6.78 (d, J=4.0 Hz, 1H), 6.48 (br s, 1H), 4.70 (d, J=6.0 Hz, 2H) ppm; 13C-NMR (125 MHz, CDCl3) δ: 166.4, 165.0 (d, 1JC–F=253.8 Hz), 142.6, 130.1 (d, 4JC–F=2.3 Hz), 129.6, 129.5 (d, 3JC–F=9.5 Hz), 126.6, 115.8 (d, 2JC–F=22.8 Hz), 112.0, 39.2 ppm; IR (ATR) 3275, 1624, 1601, 1535, 1501, 1288, 1227, 1204, 1157, 853, 768, 675, 625, 602, 536, 501, 482, 451 cm−1; HR-MS (ESI) [M+H]+ Calcd for C12H10BrFNOS: 313.9645. Found 313.9645.
Synthesis of 2,4,6-Trichlorophenyl 5-((4-Fluorobenzamido)methyl)thiophene-2-carboxylate (5)Pd(PhCN)2Cl2 (12.2 mg, 31.8 µmol, 1 mol%), Xantphos (36.8 mg, 63.6 µmol, 2 mol%), 4 (1.00 g, 3.18 mmol), 1 (1.43 g, 6.37 mmol, 2.0 eq), and toluene (6.4 mL) were added in a 50 mL round-bottomed flask equipped with an argon balloon. The flask was evacuated and backfilled with Ar three times. DBU (0.95 mL, 6.37 mmol, 2.0 eq) was added to the mixture through the septum. The flask was warmed to 80°C in an oil bath, and the mixture was stirred for 24 h. After cooling to r.t., the mixture was quenched with aq. citric acid (10% w/v), diluted with CH2Cl2, extracted with CH2Cl2 from the aqueous layer, washed with H2O, aq. Na2CO3 (0.5 M), and brine, dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by column chromatography (SiO2, hexane–EtOAc 2 : 1) to afford 5 (1.35 g, 2.94 mmol, 93%) as a white solid. mp 157.5–158.4°C; 1H-NMR (400 MHz, CDCl3) δ: 7.90 (d, J=4.0 Hz, 1H), 7.83–7.80 (m, 2H), 7.39 (s, 2H), 7.13–7.09 (m, 3H), 6.77 (br s, 1H), 4.84 (d, J=6.0 Hz, 2H) ppm; 13C-NMR (125 MHz, CDCl3) δ: 166.5, 165.1 (d, 1JC–F=253.8 Hz), 158.2, 151.6, 142.8, 136.2, 132.4, 129.94, 129.85 (d, 4JC–F=2.4 Hz), 129.7, 129.6 (d, 3JC–F=9.6 Hz), 128.7, 127.0, 115.9 (d, 2JC–F=21.5 Hz), 39.2 ppm; IR (ATR) 3316, 3090, 1748, 1641, 1541, 1499, 1445, 1335, 1254, 1229, 1042, 1013, 851, 816, 766, 729, 505 cm−1; HR-MS (DART) [M+H]+ Calcd for C19H12Cl3FNO3S: 457.9588. Found 457.9578.
Synthesis of (S)-O-Benzyl-α-alaninehydroxamic Acid TFA SaltTFA (14.6 mL) was added dropwise to a solution of (S)-N-tert-butoxycarbonyl-O-benzyl-α-alaninehydroxamate33) (1.94 g, 6.79 mmol) in CH2Cl2 (14.6 mL) at 0°C. The reaction was stirred at 0°C for 15 min and then at r.t. for 45 min. The solvents were removed in vacuo. Since the residue did not solidify, the residue was dissolved in CHCl3 (120 mL) and the solvent was removed in vacuo again to yield the desired salt as a white solid (1.96 g, 6.36 mmol, 94%). mp 96.1–98.3°C, 1H-NMR (500 MHz, D2O) δ: 7.32–7.28 (m, 5H), 4.77 (d, J=11.5 Hz, 1H), 4.73 (d, J=11.5 Hz, 1H), 3.74 (q, J=7.5 Hz, 1H), 1.24 (d, J=7.0 Hz, 3H) ppm; 13C-NMR (125 MHz, D2O) δ: 167.4, 162.9 (q, 2JC–F=35.8 Hz), 134.3, 129.9, 129.3, 128.7, 116.4 (q, 1JC–F=289.8 Hz), 78.4, 47.0, 16.5 ppm; IR (ATR) 3096, 3015, 2995, 1695, 1667, 1522, 1431, 1198, 1177, 1134, 1053, 1009, 949, 839, 793, 733, 721, 694, 615 cm−1; MS (DART) [M−CF3CO2]+ Calcd for C10H15N2O2: 195.1134. Found 195.1131; [α]D25 −36.5 (c=1.00, H2O).
Synthesis of (S)-N-(1-((Benzyloxy)amino)-1-oxopropan-2-yl)-5-((4-fluorobenzamido)methyl)-thiophene-2-carboxamide (6)(S)-O-Benzyl-α-alaninehydroxamic acid TFA salt (46.2 mg, 0.150 mmol, 1.5 eq) and DMAP (0.6 mg, 5.00 µmol, 0.05 eq) were added to a solution of 5 (45.9 mg, 0.100 mmol) and NEt3 (49 µL, 0.352 mmol, 3.5 eq) in THF (0.3 mL). The mixture was warmed to 55°C, stirred for 44 h, and cooled to r.t. The mixture was diluted with EtOAc and aq. HCl (1 M), extracted with EtOAc from the aqueous layer, washed with brine, dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by preparative TLC (SiO2, CHCl3–MeOH–AcOH=100 : 10 : 1) to afford 6 (41.8 mg, 0.0919 mmol, 92%) as a white solid. mp 185.0–185.6°C; 1H-NMR (500 MHz, CD3OD) δ: 7.91–7.88 (m, 2H), 7.63 (d, J=3.5 Hz, 1H), 7.41–7.40 (m, 2H), 7.34–7.29 (m, 3H), 7.21–7.18 (m, 2H), 7.04 (d, J=4.0 Hz, 1H), 4.83 (s, 2H), 4.72 (s, 2H), 4.36 (q, J=7.5 Hz, 1H), 1.38 (d, J=7.5 Hz, 3H) ppm; 13C-NMR (125 MHz, CD3OD) δ: 172.0, 168.8, 166.3 (d, 1JC–F=249.1 Hz), 164.1, 149.6, 138.5, 136.8, 131.6, 131.0 (d, 3JC–F=9.5 Hz), 130.6, 130.2, 129.7, 129.4, 127.3, 116.5 (d, 2JC–F=22.8 Hz), 79.0, 39.6, 17.9 ppm (one carbon signal is missing); IR (ATR) 3260, 1668, 1639, 1612, 1537, 1522, 1503, 1285, 1229, 1157, 847, 737, 694, 669, 625, 606, 557, 530, 486, 457 cm−1; MS (ESI) [M+Na]+ Calcd for C23H22FN3NaO4S: 478.1207; found 478.1200; [α]D29 −10.1 (c=0.386, MeOH).
Synthesis of (S)-5-((4-Fluorobenzamido)methyl)-N-(1-(hydroxyamino)-1-oxopropan-2-yl)thiophene-2-carboxamide (7)31)Compound 6 (34.3 mg, 0.0753 mmol) was dissolved in 1.5 mL of CH2Cl2 and cooled to 0°C in an ice bath. A solution of BCl3 in CH2Cl2 (1 M, 0.52 mL, 0.52 mmol, 6.9 eq) was added dropwise. The reaction mixture was stirred for 1 h at 0°C. The mixture was then quenched with aq. HCl (1 M) and extracted three times with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by preparative TLC (SiO2, CHCl3–MeOH 10 : 1) and by filtering through a membrane filter (0.45 µm pore size) to afford 7 (13.6 mg, 0.037 mmol, 49%) as a white solid. mp 106.0–107.4°C; 1H-NMR (500 MHz, DMSO-d6) δ: 10.65 (br s, 1H), 9.24 (t, J=5.5 Hz, 1H), 8.82 (bs, 1H), 8.48 (d, J=8.0 Hz, 1H), 7.96–7.93 (m, 2H), 7.72 (d, J=4.0 Hz, 1H), 7.31 (t, J=8.8 Hz, 2H), 7.02 (d, J=3.5 Hz, 1H), 4.61 (d, J=5.5 Hz, 2H), 4.34–4.31 (m, 1H), 1.28 (d, J=7.5 Hz, 3H) ppm; 13C-NMR (125 MHz, DMSO-d6) δ: 169.1, 165.2, 164.0 (d, 1JC–F=246.9 Hz), 160.8, 148.0, 138.1, 130.4 (2 signals), 130.0 (d, 3JC–F=8.4 Hz), 128.5, 125.9, 115.4 (d, 2JC–F=21.5 Hz), 46.6, 38.2, 18.2 ppm; IR (ATR) 3250, 3213, 3071, 2924, 1624, 1602, 1541, 1499, 1609, 1287, 1231, 1159, 1026, 849, 820, 766, 623 cm−1; MS (ESI) [M+Na]+ Calcd for C16H16FN3NaO4S: 388.0743. Found 388.0757; [α]D25 +4.4 (c=0.25, MeOH).
This study was partly supported by JSPS KAKENHI Grant Numbers 15H04634 and 15K18834, the Uehara Memorial Foundation, the Takeda Science Foundation, the Tokyo Biochemical Research Foundation, and the Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and Japan Agency for Medical Research and Development (AMED).
The authors declare no conflict of interest.
The online version of this article contains supplementary materials (copies of NMR spectra of newly obtained compounds).