1. Lycopodium Alkaloids
Since the isolation of lycopodine (5) from Lycopodium complanatum (Lycopodiaceae) by Bödeker in 1881, chemical investigations of Lycopodium plants have been actively conducted by many researchers, and nearly 300 alkaloids with diverse structures have been found.8–11) Among the Lycopodium alkaloids, huperzine A (6), which was isolated from Lycopodium serratum Thunb. in 1986, has been shown to exhibit potent acetylcholine esterase inhibitory activity and to improve memory disorders in Alzheimer’s disease patients. Other useful biological activities of Lycopodium alkaloids have also been attracting researchers in natural product chemistry, synthetic chemistry, and medicinal chemistry. In particular, the highly diverse and unique skeletal characteristics of Lycopodium alkaloids have inspired many organic chemists to make attempts at the total syntheses of these alkaloids.12–15)
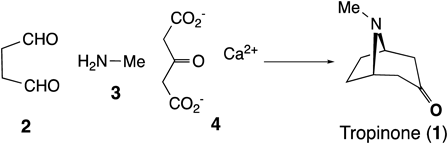
1.1. Total Syntheses of Lycodine and FlabellidineAlthough the detailed biosynthetic route to Lycopodium alkaloids remains unknown, it was demonstrated by feeding experiments that the biosynthetic precursors of lycopodine (5) are pelletierine (9) and 4-(2-piperidyl)acetoacetate (10), both of which are derived from L-lysine (8)16) (Chart 2). In this hypothetical route (Chart 2-A), we were interested in the tandem cyclization of dienamine 11A into tetracyclic 12A, which involves the formation of two bonds, i.e., between C7–C12 and C4–C13, to construct lycodine skeleton 12A from which lycopodine (5) and other representative Lycopodium alkaloids, lycodine (7) and huperzine A (6), would be biosynthesized. Taking the stereochemistry into account, the tandem cyclization of 11A into 12A would be refined to the elaborated mode as the cyclization of 11B into 12B described in Chart 2-B. Thus-formed tetracyclic intermediate 12B would be metabolized into flabellidine (13) and lycodine (7) via removal of the two hydroxy groups at C7 and C15 followed by monoacetylation or aromatization at A-ring, respectively. Inspired by this plausible hypothesis, we conceived an alternative cascade cyclization participated by ene-iminium intermediate 15 (Chart 3), which could be regarded as the quasi-equivalent of dienamine 11B.
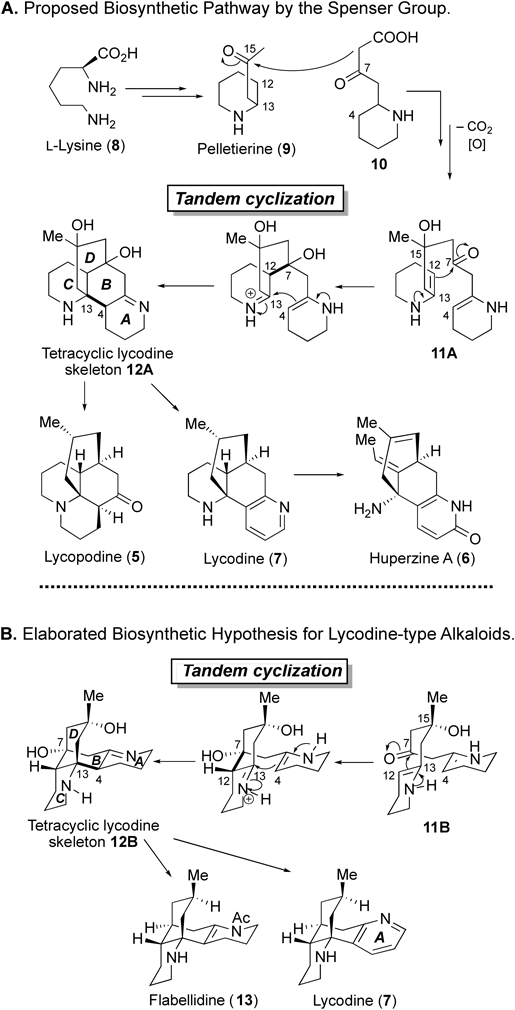
Chart 2. Hypothetical Biosynthetic Pathway for Lycopodium Alkaloids

Chart 3. Synthetic Plan Inspired by Hypothetical Biosynthesis
As shown in Chart 3, we had expected that the stereochemistry at C15 would control the stereochemical course of the conjugate addition (from 15 to 16) as well as the Mannich-like reaction (from 16 to 17) to form a tetracyclic compound that has the same stereochemistry at C7, C12, and C13 as natural lycodine-type alkaloids. Furthermore, we anticipated that ene-iminium intermediate 15 would be generated by the removal of the tert-butyloxycarbonyl (Boc) group in linear diketone 14. Using this strategy, we synthesized (−)-lycodine (7) and (+)-flabellidine (13), as follows.
Our synthesis commenced with the preparation of linear substrate 25 (Chart 4). The stereoselective construction of a methyl group at C15 was conducted by the diastereoselective Hosomi–Sakurai allylation of commercially available crotonamide 18 to give 19 in high yield and good diastereoselectivity. Upon ozonolysis of the terminal olefin in 19, the resultant aldehyde in 20 was condensed with the enolate of ethyl acetate to give lactone 21. Treatment of 21 with trimethylaluminum and N,O-dimethylhydroxylamine, followed by triethylsilyl (TES) protection, furnished di-Weinreb amide 22. The coupling reaction of 22 with alkynyl anion prepared from 23 gave di-alkynylketone derivative 24, which was subjected to hydrogenation, followed by dehydration to give linear enone 25.

Chart 4. Synthesis of Linear Substrate for Cascade Cyclization
With linear enone 25 in hand, we examined the proposed bioinspired strategy for the construction of the tetracyclic lycodine skeleton (Chart 5). After numerous surveys of the reaction conditions, we found that exposure of 25 to an excess amount of (+)-10-camphorsulfonic acid (CSA) (20 equiv, CH2Cl2, 50°C, 1 h) yielded tetracyclic lycodine skeleton 27a as the major product, probably via ene-iminium intermediate 26a. In this reaction, diastereomeric 27b was also produced (total chemical yield 61%; 27a : 27b = 3.0 : 1). Reaction intermediate 26a, which has a chair-like conformation with an equatorial methyl group at C15, would be more stable than alternative intermediate 26b, which possesses an axial methyl group, resulting in the predominant formation of 27a. The structures of 27a and 27b were elucidated by X-ray crystallographic analysis after conversion into para-bromobenzamide derivative 28 and benzamide derivative 29, respectively.

Chart 5. Bioinspired Cascade Cyclization and Structure Elucidation of the Products
For the completion of the total syntheses of 7 and 13, a mixture of 27a and 27b was subjected to debenzylation and simultaneous Boc protection to give easily separable di-Boc compounds 30 and 31 (Chart 6). The removal of Boc groups of the amines in 30 and the chemoselective acetylation exclusively gave (+)-flabellidine (13), which resulted in the first total synthesis of this alkaloid. Meanwhile, selective oxidation of A-ring with 2-iodoxybenzoic acid (IBX) furnished (−)-lycodine (7).
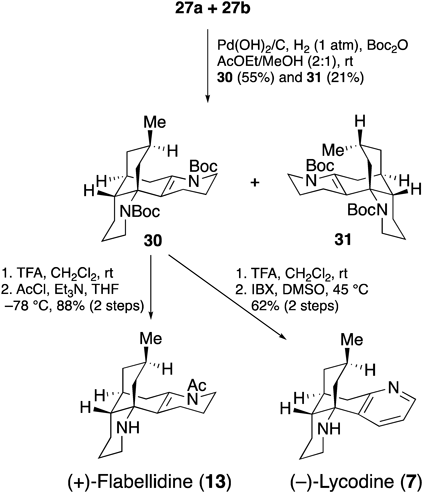
Chart 6. Syntheses of (+)-Flabellidine and (−)-Lycodine
As described above, based on the strategy inspired by biosynthesis, we have accomplished the first asymmetric total synthesis of (+)-flabellidine (13) (in 11 steps and 20.7% overall yield starting from commercially available 18), and completed the shortest total synthesis of (−)-lycodine (7) (in 11 steps and 14.6% overall yield starting from 18).17)
1.2. Total Syntheses of Lycopodine and FlabelliformineWe next planned the total synthesis of Lycopodine-type alkaloids by expanding the bioinspired strategy developed above. We had expected a similar cascade reaction starting from linear substrate 32, in which the nitrogen atom on the right end of compound 25 is replaced with an oxygen atom, to produce tetracyclic compound 34 (Chart 7). The dihydropyran ring in 34 would be incorporated by the conjugate addition of oxonium intermediate 33. Thus-obtained tetracyclic compound 34 would be directly converted into lycopodine (5).

Chart 7. Synthetic Plan for Lycopodine
Our synthesis began with the preparation of linear substrate 32 (Chart 8) for realizing the cascade cyclization reaction. Removal of Evans’s oxazolidinone moiety in 19 with lithium thiolate gave thioester 35 in 88% yield. Coupling of 35 with iodide 36 in the presence of Zn and 5 mol% PdCl2(PPh3)2 gave ketone 37 in good yield. On the other hand, enone 40 was also prepared by coupling with thioester 38 and iodide 39. Thus-obtained ketone 37 and enone 40 were subjected to olefin cross metathesis reaction using a second-generation Hoveyda–Grubbs catalyst to give desired linear substrate 32 in good yield.
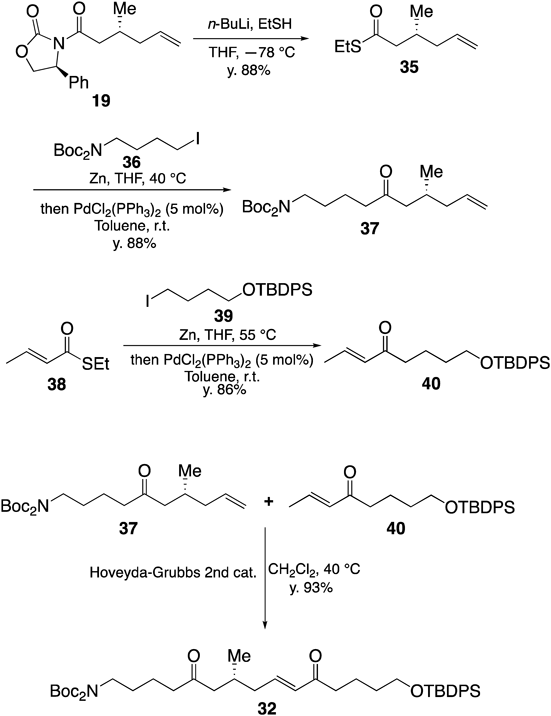
Chart 8. Synthesis of Linear Substrate 32
With linear substrate 32 in hand, we examined the reaction conditions for the key cascade cyclization step (Chart 9). Although the chemical yield needed improvement, we were able to obtain desired compound 34a in 11% yield together with diastereomeric isomer 34b in 28% yield, when 32 was treated with methanesulfonic acid in methanol at 40°C. The structures of 34a and 34b were elucidated by X-ray crystallographic analysis after conversion into 3,5-dinitrobenzamide derivatives 41a and 41b, respectively.

Chart 9. Cascade Cyclization of 32 and Structure Elucidation of the Products
Starting from tetracyclic intermediate 34a, we completed the total syntheses of lycopodine (5) and flabelliformine (42), as follows (Chart 10). Referring to Heathcock’s work,18) treatment of 34a with HBr in AcOH at room temperature (r.t.) gave the ammonium bromide salt, which was then basified with NaOH to give lycopodine (5) in 80% yield by pyrrolidine ring formation. Further, α-hydroxylation of the ketone in 5 gave flabelliformine (42) in 46% yield, thereby realizing the first asymmetric total synthesis of this alkaloid.19)

Chart 10. Syntheses of Lycopodine and Flabelliformine
The Lycopodium alkaloids synthesized in our laboratory, other than those mentioned above, are illustrated in Chart 11. They are classified according to hypothetical biogenesis into the Fawcettimine-type (fawcettimine,20) fawcettidine,20) lycoposerramine-B,21) lycoposerramine-C,22) phlegmariurine-A,22) huperzine-Q,23) and lycopoclavamine-A24)), the Phlegmarine-type (lycoposerramine-V,25) lycoposerramine-W,25) lycoposerramine-X,26) and lycoposerramine-Z26)), the Lycodine-type (lycoposerramine-R27,28)), and the Quinolizidine-type (cermizine D,29) cernuine,29) cermizine,30) and senepodine G30)). In those syntheses, consideration of the biogenetic relationships for each alkaloid type was quite instructive because the absolute configurations could be predicted when planning the asymmetric total syntheses of those alkaloids.

Chart 11. Other Lycopodium Alkaloids Synthesized in Our Laboratory
2. Monoterpenoid Indole Alkaloids
Strictosidine (45), which is biosynthesized by the condensation of tryptamine (43) and secologanin (44), is the key intermediate for the in vivo elaboration of numerous groups of monoterpenoid indole alkaloids having rich structural diversity.31) According to their structural characteristics, monoterpenoid indole alkaloids are classified into four major types (with many subdivisions), i.e., Corynanthe, Strychnos, Aspidosperma, and Iboga. (Chart 12) Biogenetically, the Corynanthe skeleton represented by geissoschizine (46) is the first compound to be formed from strictosidine (45). The skeletal changes in the monoterpene part of Strychnos alkaloids have resulted in many alkaloids classified into Aspidosperma and Iboga types. In this section, our biomimetic total syntheses of some monoterpenoid indole alkaloids related to Corynanthe and Aspidosperma types will be described.

Chart 12. Biosynthetic Relationships and Skeletons of Four Main Types of Monoterpenoid Indole Alkaloids
C-Mavacurine-type alkaloids, such as pleiocarpamine (47), normavacurine (48), and C-mavacurine (49), are considered to be biogenetically derived from geissoschizine (46) by the ring closure between C16 and N1.32,33) Geissoschizine (46) is also supposed to be a common biogenetic intermediate to provide Akuammiline-type alkaloids represented by rhazimal (50) (Chart 13). There are no reports of the total syntheses of C-Mavacurine type natural products by the direct coupling of C16 and N1 in the Corynanthe skeleton, probably because its highly strained property owing to its pentacyclic framework has made synthesis difficult.

Chart 13. Hypothetical Biosynthetic Pathway of C-Mavacurine- and Akuammiline-Type Alkaloids
Our initial synthetic plan is shown in Chart 14. We considered that the ring closure between C16 and C7 and/or C16 and N1 would occur if an electrophilic carbon species is generated at C16 in a Corynanthe-type substrate. For this purpose, we designed diazo compound 52, which was prepared from geissoschizine (46) through the Regitz diazo transfer reaction.

Chart 14. Initial Synthetic Plan for C-Mavacurine-Type Alkaloids
Our synthesis commenced with the preparation of diazo compound 52 via (±)-46, as shown in Chart 15. According to Vincent’s report,34) α,β-unsaturated ester 55 was prepared from tryptamine (43). The Ni(cod)2-mediated reductive cyclization of 55 afforded trans-56 as the major product in 55% yield together with desired cis-56 in 31% yield. Then, we examined the conversion of trans-56 into cis-56. After several attempts, we found that treatment of iminium 57 with Noyori catalyst and formic acid/Et3N in N,N-dimethylformamide (DMF) in the presence of titanium tetraisopropoxide at 30°C gave cis-56 predominantly (cis : trans = 5.3 : 1). Thus obtained cis-56 was treated with excess lithium diisopropylamide (LDA) and then with methyl formate to provide (±)-geissoschizine (46) in 80% yield. Next, (±)-46 was converted into diazo compound 52 in 60% yield by employing tosyl azide in the presence of Et3N.

Chart 15. Synthesis of Cyclization Precursor Diazo Compound 52
With diazo compound 52 in hand, we next investigated the cyclization under carbene-generating conditions. Among the binuclear rhodium complex catalysts used, a separable product was obtained in 54% yield only when Rh2(OAc)4 was employed as the catalyst. However, the product was unexpected structure 58 generated by D-ring expansion. The use of mononuclear rhodium complex (PPh3)3RhCl or copper catalyst Cu(MeCN)·BF4 did not promote the reaction at room temperature. When JohnPhosAu-(MeCN)SbF6 was used as the catalyst, ring-expanded product 58 was again obtained in 36% yield together with β-hydride elimination product 59 (36% yield). The ring expansion giving 58 would proceed by the 1,2-alkyl migration of the C20 ethylidene side chain onto the metal carbenoid at C16, as illustrated in Chart 16.

Chart 16. A Plausible Mechanism of the Unexpected Ring Expansion
Given the above results, we reformulated our synthetic plan by taking the molecular conformation of Corynanthe-type alkaloids into consideration. Normal-type Corynanthe alkaloids, such as geissoschizine (46), are known to take the trans-quinolizidine form (46a),35) but can likewise adopt the cis-quinolizidine conformer (46b) through the inversion of the lone electron pair of nitrogen atom (N4) (Chart 17). We anticipated that when compound 52 took the cis-quinolizidine form, the distance of the reaction sites (between C16 and C7 and/or C16 and N1) was reduced, effecting the desired cyclization. Then, we attempted to modify the lone electron pair of N4 in compound 52 so that 52 would take the cis-quinolizidine form. We chose N4-borane complex (60) and prepared it by treatment of 52 with the BH3–tetrahydrofuran (THF) complex (Chart 18). Nuclear Overhauser effect (NOE) experiments of compound 60 (observation of signal between H-6β and H-21β) revealed that the complex had the cis-quinolizidine form, as expected (Chart 17).

Chart 17. Conformations of Geissoschizine and 60
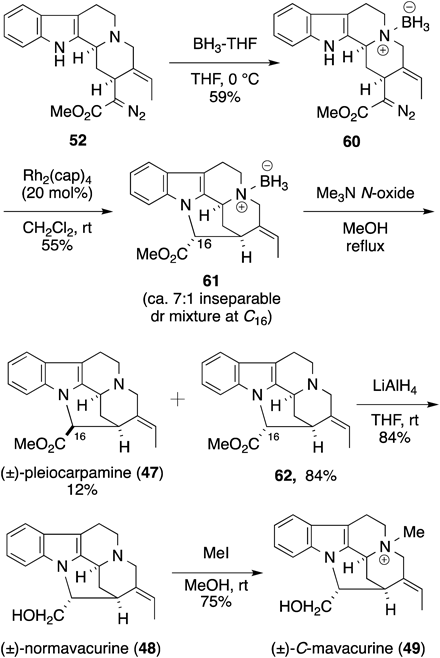
Chart 18. Synthesis of Pleiocarpamine, Normavacurine, and C-Mavacurine
With cyclization precursor 60 in hand, we examined again various metal catalysts for the cyclization. The Rh2(cap)4 catalyst gave cyclized product 61 in 55% yield as an inseparable mixture of diastereomers at C16 (approx. 7 : 1) through a carbene N–H insertion reaction. The amine–borane bond in product 61 was easily cleaved by heating 61 in the presence of trimethylamine oxide to give (±)-pleiocarpamine (47) and compound 62 (16-epi-pleiocarpamine) in 12% and 84% yields, respectively. All spectroscopic data of synthetic 47 were completely identical with reported data. Reduction of methyl ester 62 with LiAlH4 afforded (±)-normavacurine (48) in 84% yield, and 48 was further converted into (±)-C-mavacurine (49) in 75% yield by treatment with excess methyl iodide. The spectral and physical properties of synthetic 48 and 49 were in good agreement with the reported data of the natural products.
As described, we have achieved the biomimetic total syntheses of (±)-pleiocarpamine (47), (±)-normavacurine (48), and (±)-C-mavacurine (49) via a direct cyclization by a carbene N–H insertion reaction between C16 and N1 in Corynanthe compound 60. For this key cyclization, the N4 modification of the substrate using an amine-borane complex was indispensable to fix the molecular conformation to a robust cis-quinolizidine structure.36)
2.2. Total Synthesis and Structure Elucidation of Kopsiyunnanine KThe genus Kopsia, which belongs to Family Apocynaceae, is a rich source of monoterpenoid indole alkaloids possessing structural diversity as well as a wide range of biological activities.37) As a result of our continuing chemical studies on novel bioactive alkaloids, we were able to accomplish the structure elucidation of several unique monoterpenoid indole alkaloids from Kopsia arborea native to Yunnan Province in China38–46) (Chart 19).
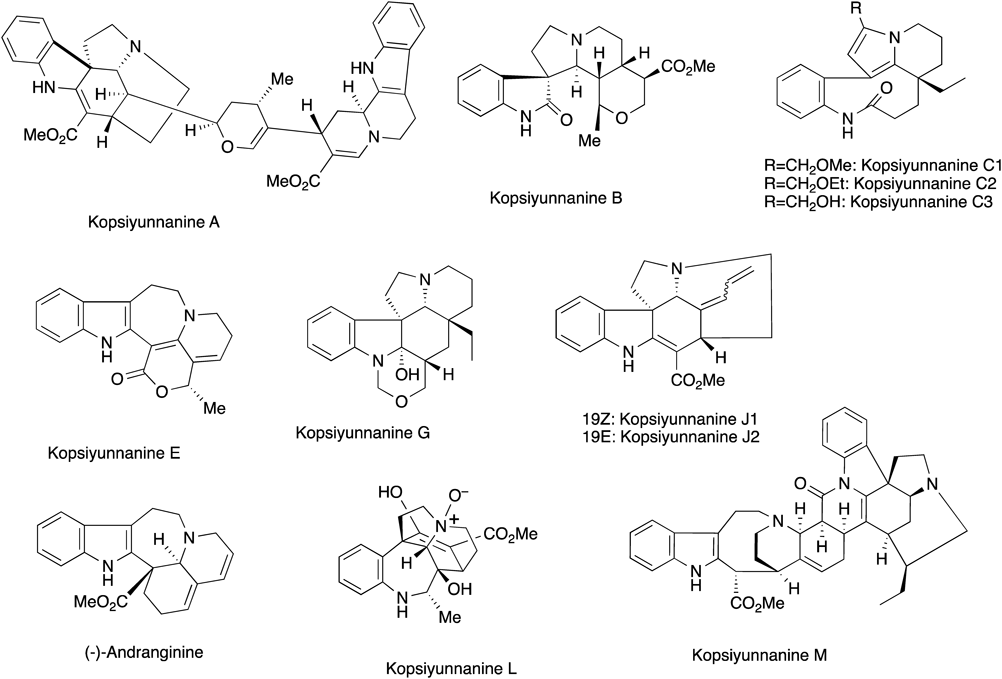
Chart 19. Representative New Alkaloids Found in Kopsia arborea
Further investigations have led to the isolation of an intriguing new alkaloid named kopsiyunnanine K (63). From spectroscopic analyses, kopsiyunnanine K was proposed to be a monoterpenoid indole alkaloid bearing an azepine-fused tetrahydro-β-carboline ring system, which has never been discovered in monoterpenoid indole alkaloids. The absolute configurations at C16 and C20 were deduced as follows. The circular dichroism (CD) spectrum of 63 showed the opposite Cotton effect to that of decarbomethoxydihydrogambirtannine (64), a Yohimbane-type alkaloid possessing 3S configuration, which led us to propose that the configuration at C16 in 63 is R (Chart 20).

Chart 20. CD Spectra of Kopsiyunnanine K and 64
The configuration at C20 was deduced from a biogenetic point of view (Chart 21). Biogenetically, 63 would be generated from an Aspidosperma-type alkaloid, vincadifformine (see also Chart 12), via (−)-eburnamine (65) that co-exists in K. arborea. Cleavage of the bond between C2 and C21 in 65 would occur by an initial protonation at C2 of the indole moiety to give C/E-ring opening intermediate A. Next, hydrolysis of the iminium function in A would give aldehyde B. By oxidation and esterification of the aldehyde function in B followed by the formation of an aldehyde group at C16 from the hemiaminal function in B, possible biogenetic precursor C of kopsiyunnanine K (63) would be generated. Then, 63 would be formed by the Pictet-Spengler reaction of C. This hypothesis allowed us to suggest that 63 has a rearranged skeleton with C20R configuration.

Chart 21. Possible Biogenetic Route to Kopsiyunnanine K
To clarify the structure as well as the absolute configuration at C16 and C20 of 63, an asymmetric total synthesis was performed (Chart 22). We initially prepared ester derivative 69 from δ-valerolactone (66) via a five-step operation: alkylation of 66 with EtI, methanolysis, methoxymethyl (MEM) protection of primary alcohol, alkaline hydrolysis of 67, and condensation of the resultant carboxylic acid with chiral alcohol 68. The asymmetric Ireland–Claisen rearrangement of MEM-protected allyl alcohol47) derivative 69 was performed under the optimum conditions using potassium hexamethyldisilazide (KHMDS) and trimethylsilylchloride (TMSCl) in toluene. Methylation of resultant carboxylic acid 70 gave ester 71 in 90% yield with 92% enantiomeric excess (ee) in 2 steps. The efficient chirality transfer process of the Ireland–Claisen rearrangement would be explained by using the transition state depicted in Chart 22. In the chair-like transition state, the phenyl group would take the pseudo-equatorial position. Furthermore, in the ketene acetal moiety, the side chain with the MEM group would favor the equatorial position with the aid of the chelate effect. Next, an amine function was introduced to 71 by deprotection of the MEM group followed by the Mitsunobu reaction with nosyl (Ns) amine to afford amide 72. After ozonolysis of 72, alkylation of the resultant amide with indole unit 73 was executed to give aldehyde 74. Deprotection of the Ns group in 74 followed by the intramolecular diastereoselective Pictet–Spengler reaction of the resultant amine in the presence of trifluoroacetic acid gave kopsiyunnanine K (63) as a single diastereomer in a quantitative yield. Recrystallization of the product afforded optically pure 63. The structure and the 16R, 20R configuration of synthetic 63 were confirmed by X-ray crystallographic analysis. Synthetic 63 was identical in all respects with the natural compound, including the optical property. Therefore, the structure and the absolute configuration of kopsiyunnanine K were established, as shown in formula 63.48)
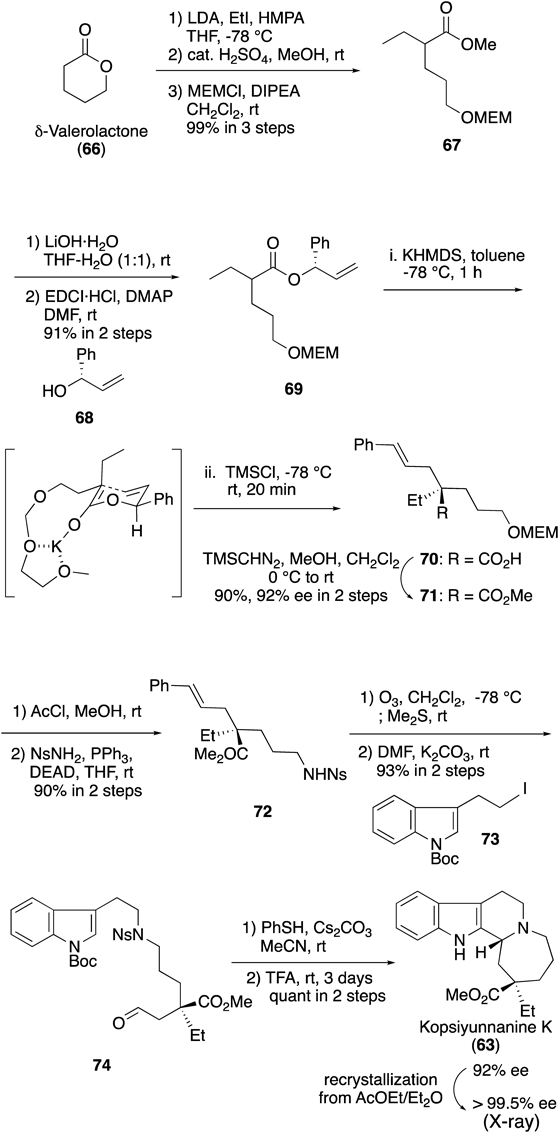
Chart 22. Synthesis of Kopsiyunnanine K
There are three plant species in the genus Gelsemium. Among them, Gelsemium elegans Benth. (Gelsemiaceae) native to Southeast Asia and southern China is a toxic plant. Nevertheless, it has been used in traditional Chinese medicine for the treatment of skin ulcers and dermatitis and as a remedy for cancer-related pain.49) In our previous study, we were able to prove that the plant of origin of ‘Yakatsu,’ an ancient medicine stored in The Shosoin Repository in Japan for over 1250 years, was G. elegans.50) Phytochemical studies of this plant by us51–54) and other groups have led to the isolation and structure elucidation of more than one hundred monoterpenoid indole alkaloids with diverse chemical structures.51) Among the six classes of Gelsemium alkaloids,51) the so-called Gelsedine-type alkaloids were found to exhibit potent cytotoxic activity against A431 epidermoid carcinoma cells.55) Further, koumine (78), one of the major constituents of this plant, was found to exhibit toxicity toward cancer cells,56) neuroprotective effects,57) and anti-inflammatory effects.58) However, due to the small quantities of the natural products, a comprehensive biological evaluation of these alkaloids could not be accomplished. Therefore, to accelerate further investigation of pharmacological activities, we embarked on the total synthesis of these classes of alkaloids. In this section, the biomimetic total syntheses of koumine (78) and 11-methoxy-19R-hydroxygelselegine (90) will be described.
A hypothetical biogenetic route to koumine (78) is shown in Chart 23. Like other monoterpenoid indole alkaloids, strictosidine (45) is considered to be the primal biogenetic precursor of this alkaloid. By the intramolecular carbon–carbon bond formation between C16 and C5 in Corynanthe-type alkaloid geissoschizine (46), E-koumidine (75) belonging to the Sarpagine-type would be formed. Then, an oxidation at the allylic position of 75 and a subsequent C/D ring cleavage would give hypothetical biogenetic precursor 77, and an intramolecular SN2′ reaction in 7759,60) would result in the coupling between C7 and C20, producing koumine (78). For the synthesis of this cage structure alkaloid, we planned to mimic this hypothetical biogenetic key reaction at the final stage of the total synthesis, and started chemical synthesis using known chiral diol derivative (79) as the starting material.

Chart 23. Hypothetical Biogenetic Route to Koumine
Starting from known chiral diol 79,61) keto-amine 80 was prepared in four steps (Chart 24). Then, 80 was alkylated with alkyne derivative 81 to afford alkyne-ketone 82. Silylenol ether 83 was prepared and subsequent gold(I)-catalyzed cyclization gave 6-exo-dig-mode cyclized product 84 having E-olefin in 85% yield.62) Thus-obtained cyclized product was converted into ketone 85 by introducing the C1 unit to C17 with the Tebbe reagent and subsequent oxidation at C3 in compound 84. By Fischer indole synthesis using ketone 85 with a phenylhydrazine derivative, indole compound 86 was obtained. The chemo- and diastereoselective introduction of a hydroxy group at C17 was achieved by hydroboration using 9-borabicyclo[3.3.1]nonane (9-BBN) to give 87, which was subjected to the Birch reduction at −78°C to afford Sarpagine-type unnatural indole alkaloid 76, which corresponds to one of the possible biogenetic intermediates of koumine (78).

Chart 24. Biomimetic Total Synthesis of Koumine -1-
Next, according to the biogenetic speculation mentioned above, the C/D ring opening in 76 was carried out with methyl chloroformate to give compound 88 (Chart 25). The carbamate was reduced to an N-methyl group and the allylic alcohol was acetylated. Finally, the intramolecular coupling reaction between C7 and C20 in 89 was achieved by palladium catalysis in the presence of sodium hydride (NaH) to furnish koumine (78) in good yield. Thus, we have achieved the total synthesis of (−)-koumine in 18 steps in a biomimetic manner.63)
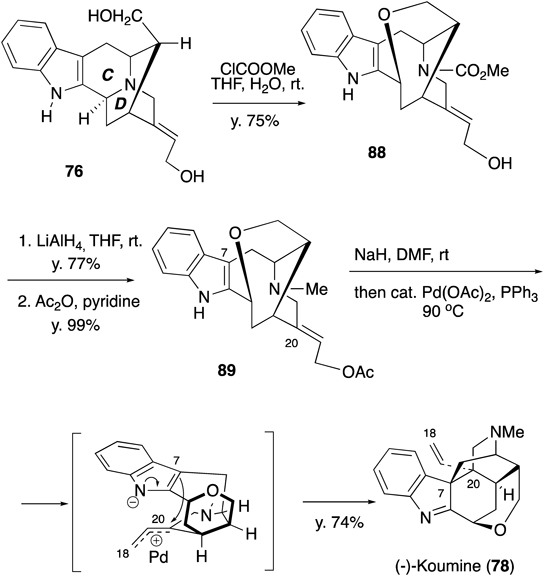
Chart 25. Biomimetic Total Synthesis of Koumine -2-
The proposed biosynthesis of 11-methoxy-19R-hydroxygelselegine (90), which has a pentacyclic oxindole structure with seven chiral centers, is shown in Chart 26. Humantenine-type oxindole alkaloid 91 would also be derived from Sarpagine-type alkaloid 75 by oxidation of the indole nucleus. Then, the C19–C20 double bond in Humantenine-type alkaloid 91 would be oxidized to form epoxy derivative 92. By a subsequent attack of the nitrogen on the epoxy carbon, aziridinium intermediate 93 would be generated. Then, the ring opening between C21 and N4 by the attack of water would give rise to alkaloid 90.64) In order to realize this biogenetic hypothesis in a chemical flask, we designed a synthesis of this alkaloid starting from compound 85, which is a synthetic intermediate of koumine (78).

Chart 26. Hypothetical Biogenetic Route to 11-Methoxy-19R-hydroxygelselegine
A synthetic intermediate of koumine was utilized as follows (Chart 27). A Fischer indole synthesis using ketone 85 and 2-methoxy-phenylhydrazine derivative in the presence of pyridine·HCl gave desired 11-methoxyindole derivative 94. The introduction of a hydroxy group at C17 was again achieved by hydroboration using 9-BBN to give 95. A Birch reduction at −30°C gave sarpagine-type natural product gardnerine (96) by the removal of benzyl and allylic hydroxy groups in 95. Next, according to biogenetic considerations, C/D ring cleavage of 96 was carried out using trichloroethyl chloroformate to afford 97. Then, treatment of 97 with two equivalents of osmium tetroxide afforded oxindole-diol 98 as the sole product via a stereoselective pinacol-type rearrangement and a simultaneous stereoselective dihydroxylation of the double bond. After installation of a methoxy group on N1 of oxindole, the diol in 99 was converted into epoxy derivative 100 by a conventional procedure. Removal of the protecting group on N4 in 100 gave secondary amine 92, which was then heated in dioxane to afford desired aziridine derivative 93. Finally, aziridine derivative 93 was treated with CF3CO2H in THF to generate natural product 90 by a ring opening between C21 and N4. Thus, we have achieved the biomimetic synthesis of structurally unique alkaloid, 11-methoxy-19R-hydroxygelselegine (90), in a highly stereoselective manner.

Chart 27. Biomimetic Synthesis of 11-Methoxy-19R-hydroxygelselegine
Further, the divergent transformation of 14-hydroxygelsenicine into biogenetically related Gelsemium alkaloids, i.e., (−)-gelsefuranidine,53) (−)-gelselegandine B,65) (−)-gelselegandine C,65) and (−)-gelsemolenine A,66) was accomplished recently.67)