Experimental
General MethodsNMR spectra were recorded on Varian 400-MR (1H-NMR at 400 MHz, 13C-NMR at 100 MHz) and Bruker AVANCE III HD-500 (1H-NMR at 500 MHz, 13C-NMR at 125 MHz) spectrometers, using tetramethylsilane (TMS) or CDCl3 as the reference standard (1H-NMR at 0.00 ppm (TMS), 13C-NMR at 77.0 ppm (CDCl3)). Chemical shifts are reported in ppm. Peak multiplicities use the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; quin, quintet; sept, septet; m, multiplet; br, broadened. IR spectra were recorded on a JASCO FT/IR-4200 spectrometer. Mass spectra and high-resolution mass spectra (HRMS) were obtained on JEOL JMS-700 and Waters SYNAPT G2-Si HDMS mass spectrometers, respectively. Melting points (mp) were recorded on Yanaco MP-500D and SRS Opti Melt MPA 100 apparatuses and were uncorrected. Analytical TLC was performed on precoated plates (0.25 mm, Merck silica gel 60 F254). Column chromatography was performed with silica gel (40–50 µm, Kanto Chemical Co., Inc., Japan). Preparative HPLC was performed on a recycling system utilizing LC-9210 II Next and LC-5060 instruments (JAI Co., Inc., Japan). Commercially available chemicals were used as purchased. Diarylamines and triarylamines were prepared according to the method reported in the literature.43)
General Procedure for Intramolecular Oxidative Coupling of Diarylamines: N-Benzyl-2,3,6,7-tetramethoxycarbazole 2a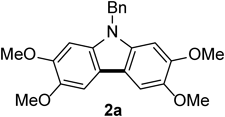
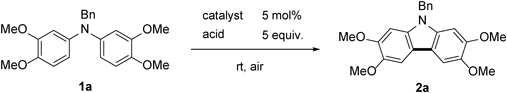
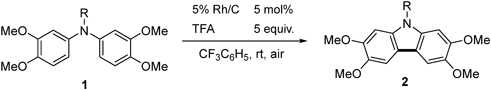
To a solution of N-benzyldiarylamine (1a, 156 mg, 0.40 mmol) and trifluoroacetic acid (0.15 mL, 2.0 mmol) in benzotrifluoride (2 mL) was added 5% Rh/C (42 mg, 0.020 mmol) and stirred at room temperature under open-air conditions for 2 h. After addition of a saturated NaHCO3 solution, the resulting mixture was extracted with AcOEt and the organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo to give the crude product, which was purified by silica gel column chromatography (30% AcOEt–hexane) to afford 142 mg (92%) of the title compound. Recrystallization from AcOEt–hexane afforded 2a as colorless needles (mp 176–178°C). 1H-NMR (500 MHz, CDCl3) δ: 7.47 (2H, s), 7.30–7.21 (3H, m), 7.14–7.08 (2H, m), 6.78 (2H, s), 5.43 (2H, s), 4.01 (6H, s), 3.89 (6H, s). 13C-NMR (125 MHz, CDCl3) δ: 148.3 (s), 144.1 (s), 137.2 (s), 135.1 (s), 128.8 (d), 127.4 (d), 126.2 (d), 115.1 (s), 102.1 (d), 93.0 (d), 56.6 (q), 56.2 (q), 46.7 (t). IR (ATR): 1473, 1242, 1152, 1049 cm−1. MS (EI) m/z: 91 (Bn), 362 (M+ − CH3), 377 (M+, 100%), 378 (M+ + H). HRMS (EI) m/z Calcd for C23H23NO4 (M+): 377.1627. Found: 377.1625.
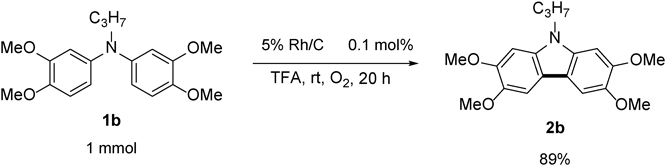
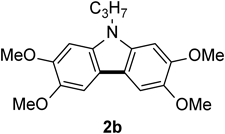
N-Propyl-2,3,6,7-tetramethoxycarbazole 2bThe title compound (2b, 111 mg, 85%) was prepared according to General procedure (conditions: 2 h) using N-propyldiarylamine (1b, 132 mg, 0.40 mmol). Pale brown prism (mp 168–170°C; AcOEt-hexane). 1H-NMR (500 MHz, CDCl3) δ: 7.44 (2H, s), 6.86 (2H, s), 4.19 (2H, t, J = 7.0 Hz), 4.00 (12H, s), 1.89 (2H, sext, J = 7.0 Hz), 098 (3H, t, J = 7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 148.2 (s), 143.8 (s), 134.9 (s), 114.9 (s), 102.1 (d), 92.8 (d), 56.6 (q), 56.3 (q), 44.8 (t), 22.4 (t), 11.8 (q). IR (ATR): 1479, 1254, 1200, 1153, 1059, 839 cm−1. MS (EI) m/z: 314 (M+ − CH3), 329 (M+, 100%), 330 (M+ + H). HRMS (EI) m/z Calcd for C19H23NO4 (M+): 329.1627. Found: 329.1620.
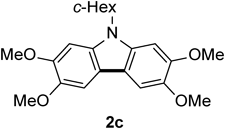
N-Cyclohexyl-2,3,6,7-tetramethoxycarbazole 2cThe title compound (2c, 146 mg, 98%) was prepared according to General procedure (conditions: 6 h) using N-cyclohexyldiarylamine (1c, 148 mg, 0.40 mmol). Brown solids (mp 190–191°C; CHCl3-hexane). 1H-NMR (500 MHz, CDCl3) δ: 7.42 (2H, s), 7.01 (2H, s), 4.35–4.26 (1H, m), 4.01 (6H, s), 4.00 (6H, s), 2.35–2.25 (2H, m), 2.05–1.84 (5H, m), 1.61–1.51 (2H, m), 1.42–1.31 (1H, m). 13C-NMR (125 MHz, CDCl3) δ: 147.8 (s), 143.8 (s), 134.1 (s), 115.6 (s), 101.9 (d), 94.7 (d), 56.6 (q), 56.5 (q), 55.5 (d), 31.0 (t), 26.5 (t), 25.8 (t). IR (ATR): 1477, 1155, 1064, 836 cm−1. MS (EI) m/z: 354 (M+ − CH3), 369 (M+, 100%), 370 (M+ + H). HRMS (EI) m/z Calcd for C22H27NO4 (M+): 369.1940. Found: 369.1944.
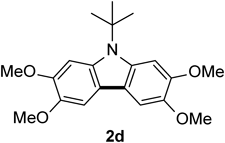
N-tert-Butyl-2,3,6,7-tetramethoxycarbazole 2dThe title compound (2d, 52 mg, 38%) was prepared according to General procedure (conditions: O2, 2 h) using 5% Pd/Al2O3 (43 mg, 0.020 mmol) and N-tert-butyldiarylamine (1d, 138 mg, 0.40 mmol). Pale yellow prism (mp 195–199°C; AcOEt–hexane). 1H-NMR (500 MHz, CDCl3) δ: 7.38 (2H, s), 7.34 (2H, s), 4.00 (6H, s), 3.97 (6H, s), 1.97 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 147.0 (s), 143.7 (s), 134.6 (s), 117.1 (s), 101.0 (d), 98.7 (d), 58.5 (s), 56.5 (q), 56.4 (q), 31.0 (q). IR (ATR): 1483, 1464, 1139, 1021 cm−1. MS (EI) m/z: 287 (M+ + H − tert-Bu, 100%), 343 (M+), 344 (M+ + H). HRMS (EI) m/z Calcd for C20H25NO4 (M+): 343.1784. Found: 343.1781.
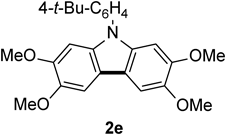
N-(4-tert-Butyphenyl)-2,3,6,7-tetramethoxycarbazole 2eThe title compound (2e, 157 mg, 94%) was prepared according to General procedure (conditions: 2 h) using triarylamine (1e, 169 mg, 0.40 mmol). Colorless needles (mp 161–164°C; AcOEt–hexane). 1H-NMR (500 MHz, CDCl3) δ: 7.61 (2H, d, J = 8.5 Hz), 7.47 (2H, d, J = 8.5 Hz), 7.46 (2H, s), 6.90 (2H, s), 4.03 (6H, s), 3.89 (6H, s), 1.44 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 150.1 (s), 148.3 (s), 144.6 (s), 135.33 (s), 135.30 (s), 126.9 (d), 126.1 (d), 115.5 (s), 101.7 (d), 93.9 (d), 56.6 (q), 56.3 (q), 34.8 (s), 31.4 (q). IR (ATR): 1487, 1470, 1194, 1120 cm−1. MS (EI) m/z: 404 (M+ − Me), 419 (M+, 100%), 420 (M+ + H). HRMS (EI) m/z Calcd for C26H29NO4 (M+): 419.2097. Found: 419.2095.
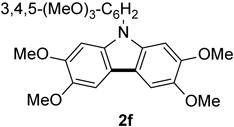
N-(3,4,5-Trimethoxyphenyl)-2,3,6,7-tetramethoxycarbazole 2fThe title compound (2f, 69 mg, 38%) was prepared according to General procedure (conditions: 3 h) using triarylamine (1f, 182 mg, 0.40 mmol). Pale pink needles (mp 147–148°C; Et2O). 1H-NMR (500 MHz, CDCl3) δ: 7.46 (2H, s), 6.89 (2H, s), 6.75 (2H, s), 4.03 (6H, s), 3.99 (3H, s), 3.90 (6H, s), 3.88 (6H, s). 13C-NMR (125 MHz, CDCl3) δ: 154.2 (s), 148.3 (s), 144.7 (s), 137.1 (s), 135.4 (s), 133.7 (s), 115.5 (s), 104.3 (d), 101.9 (d), 93.8 (d), 61.0 (q), 56.7 (q), 56.33 (q), 56.29 (q). IR (ATR): 1597, 1474, 1451, 1426, 1160, 1124, 1041, 1016, 994 cm−1. MS (EI) m/z: 438 (M+ − Me), 453 (M+, 100%), 454 (M+ + H). HRMS (EI) m/z Calcd for C25H27NO7 (M+): 453.1788. Found: 453.1795.
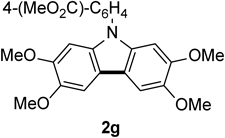
N-(4-Methoxycarbonylphenyl)-2,3,6,7-tetramethoxycarbazole 2gThe title compound (2g, 136 mg, 81%) was prepared according to General procedure (conditions: O2, 21 h) using triarylamine (1g, 168 mg, 0.40 mmol). Pale brown needles (mp 226–227°C; AcOEt). 1H-NMR (500 MHz, CDCl3) δ: 8.30 (2H, d, J = 8.5 Hz), 7.66 (2H, d, J = 8.5 Hz), 7.45 (2H, s), 6.94 (2H, s), 4.03 (6H, s), 3.99 (3H, s), 3.88 (6H, s). 13C-NMR (125 MHz, CDCl3) δ: 166.4 (s), 148.4 (s), 145.1 (s), 142.5 (s), 134.5 (s), 131.5 (d), 128.4 (s), 126.0 (d), 116.2 (s), 101.8 (d), 93.8 (d), 56.6 (q), 56.3 (q), 52.4 (q). IR (ATR): 1719, 1606, 1280, 1198, 1166, 1106 cm−1. MS (EI) m/z: 406 (M+ − Me), 421 (M+, 100%), 422 (M+ + H). HRMS (EI) m/z Calcd for C24H23NO6 (M+): 421.1525. Found: 421.1534.
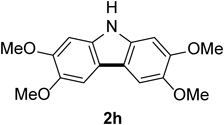
2,3,6,7-Tetramethoxycarbazole 2hColorless needles (mp 231–233°C; AcOEt). 1H-NMR (500 MHz, CDCl3) δ: 7.73 (1H, br s), 7.40 (2H, s), 6.93 (2H, s), 4.00 (6H, s), 3.96 (6H, s). 13C-NMR (125 MHz, CDCl3) δ: 148.2 (s), 144.3 (s), 134.0 (s), 115.8 (s), 101.9 (d), 94.5 (d), 56.6 (q), 56.2 (q). IR (ATR): 3400, 1491, 1473, 1196, 1128, 1001, 838 cm−1. MS (EI) m/z: 272 (M+ − Me), 287 (M+, 100%), 288 (M+ + H). HRMS (EI) m/z Calcd for C16H17NO4 (M+): 287.1158. Found: 287.1151.
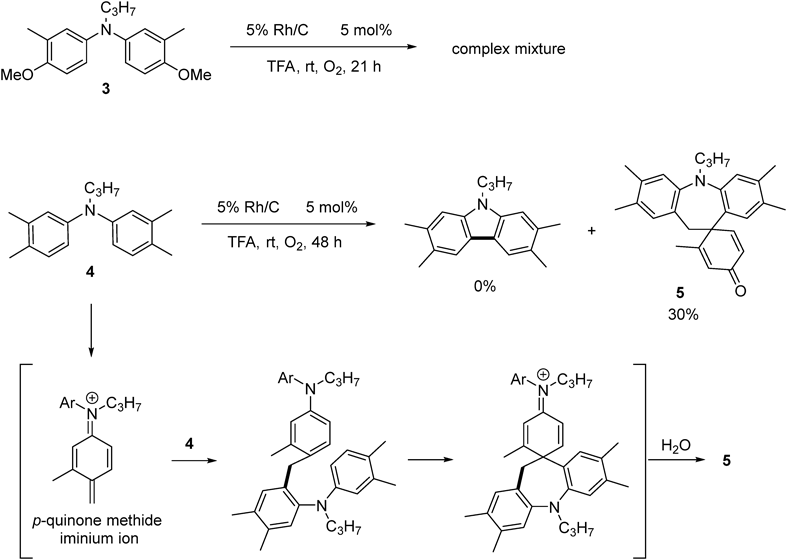
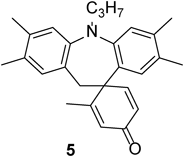
N-Propyldihydrodibenzoazepine 5The title compound (5, 11 mg, 30%) was prepared according to General procedure (conditions: O2, 48 h) using diarylamine (4, 54 mg, 0.20 mmol). Pale brown oil. 1H-NMR (500 MHz, CDCl3) δ: 6.93 (1H, s), 6.90 (1H, s), 6.88 (1H, s), 6.79 (1H, d, J = 10 Hz), 6.39 (1H, s), 6.18 (1H, d, J = 10 Hz), 6.14 (1H, s), 3.74–3.62 (2H, m), 2.20 (3H, s), 2.19 (6H, s), 2.01 (3H, s), 1.88 (3H, s), 1.61 (2H, sext, J = 7.5 Hz), 0.95 (3H, t, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ: 186.7 (s), 166.4 (s), 157.0 (d), 148.5 (s), 145.4 (s), 136.5 (s), 135.8 (s), 132.1 (d), 131.9 (s), 130.7 (d), 130.2 (s), 130.1 (s), 126.1 (d), 124.5 (d), 121.7 (d), 120.4 (d), 52.0 (t), 50.9 (s), 40.0 (t), 21.2 (t), 20.5 (q), 19.8 (q), 19.7 (q), 19.2 (q), 18.6 (q), 11.9 (q). IR (ATR): 1661, 1498, 879, 730 cm−1. MS (EI) m/z: 342 (M+ − C3H7), 356 (M+ − C2H5), 385 (M+, 100%). HRMS (EI) m/z Calcd for C27H31NO (M+): 385.2406. Found: 385.2410.