Review
Development of Copper-Catalyzed Chemoselective Reactions
2020 Volume 68 Issue 5 Pages 405-420
Details
2020 Volume 68 Issue 5 Pages 405-420
Chemoselective reactions can contribute to streamlining synthesis of pharmaceuticals, agrochemicals, and other functional molecules by avoiding use of protecting groups. In this review, copper catalysts were demonstrated useful for developing two types of chemoselective reactions: C–C bond-forming reactions at an anomeric carbon of unprotected aldoses and difunctionalization reaction of C–C multiple bonds. The “soft” nucleophilic copper species exhibit orthogonal reactivity toward “hard” polar functional groups and preferentially react with “soft” functional groups. The catalysis also controls stereoselectivity and/or regioselectivity to provide value-added products from readily available feedstock compounds.
Biologically active molecules often possess multiple functional groups. Synthesis of these compounds requires protecting groups to secure the promotion of intended transformation at the particular functional group of the starting molecule. The protecting groups, however, are not included in the final products. Hence the use of protecting groups diminishes the synthetic efficiency in terms of atom-1) and step-economy.2) In this circumstance, development of chemoselective reactions, which refers to selective promotion of one particular reaction over other possible reaction pathways, is of benefit to develop pharmaceuticals, agrochemicals and other functional molecules in several aspects (Fig. 1). Avoiding the protection and deprotection sequences, at least, decreases the number of synthetic steps by two, streamlining the overall synthesis. It also results in decrease of waste generation derived from protecting groups. Furthermore, chemoselective reactions can be applied to direct modification of biologically active compounds, such as drug leads and natural products, which is considered as late-stage diversification approach.3–5)

Hard and soft acids and bases (HSAB) theory6,7) is a useful guideline for developing chemoselective reactions. The HSAB theory classifies Lewis acids and Lewis bases into “hard” or “soft” depending on the nature of the compounds. The reactivity between them can be simply summarized as follows: a reaction between soft acids and soft bases or hard acids and hard bases is faster and provides stronger bonding than other combinations. Based on this fundamental principle, we focused our attention on the use of copper-conjugated soft carbon nucleophiles to develop chemoselective C–C bond-forming reactions. The soft organocopper species chemoselectively reacts with soft carbon electrophiles in the presence of polar hard functional groups without protecting these possible reactive sites. In this review we introduce two types of transformations: C–C bond-forming reactions at anomeric position of unprotected aldoses and difunctionalizations of C–C multiple bonds.
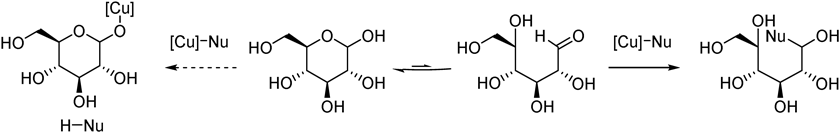
Sugars are highly important molecules in biological systems and attractive resources for drug lead compounds.8–10) The transformation of sugars, however, usually requires tedious protection/deprotection of multiple hydroxy groups to control reaction pathways, especially when carbon elongation reaction is conducted.11) Although direct use of unprotected aldoses for carbon elongation reaction is desirable in terms of efficiency, protonolysis of organometallic reagents by free hydroxy groups is a severe competitive side reaction (Fig. 2). Hence the development of carbon elongation reactions that are compatible with free hydroxy groups has been highly demanded to circumvent the protection/deprotection sequences.12–15) Our working hypothesis to tackle this issue is soft–hard orthogonal reactivity between soft organocopper species and hard hydroxy groups: soft organocopper species preferentially reacts with a carbon electrophile through predominant orbital interaction while ionic protonation pathway is suppressed. Based on this hypothesis, we could add the following three new reactions to the collection of C–C bond-forming reactions of unprotected aldoses.
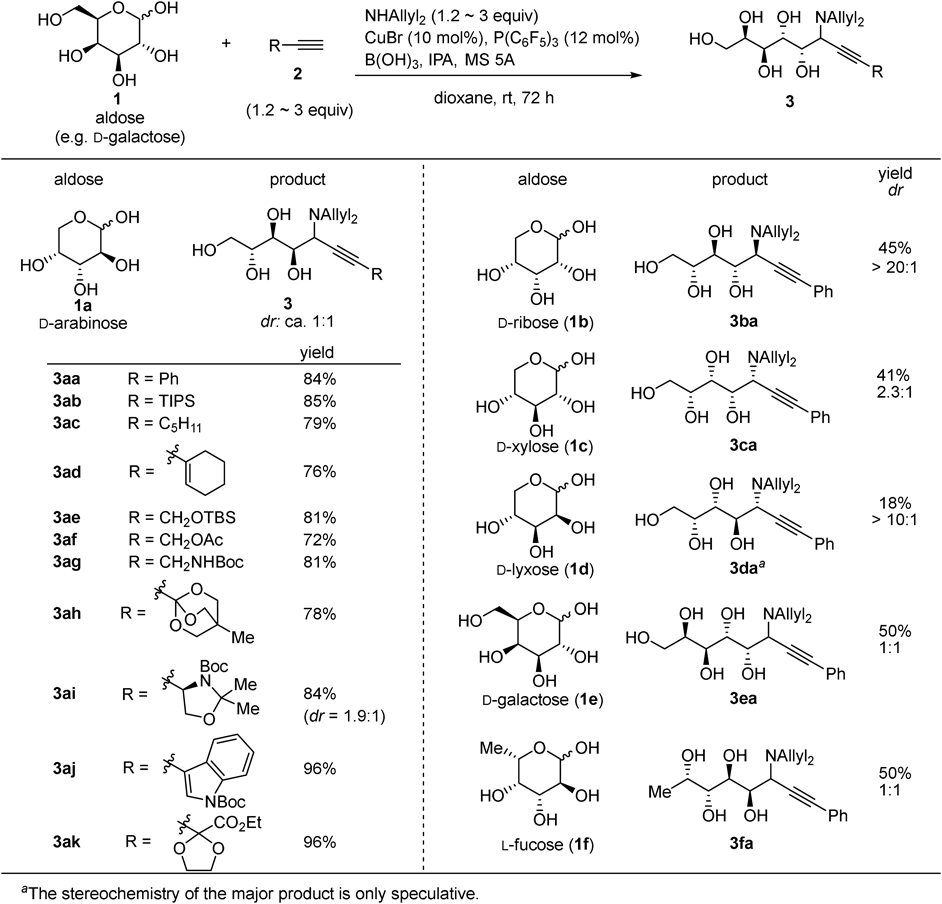
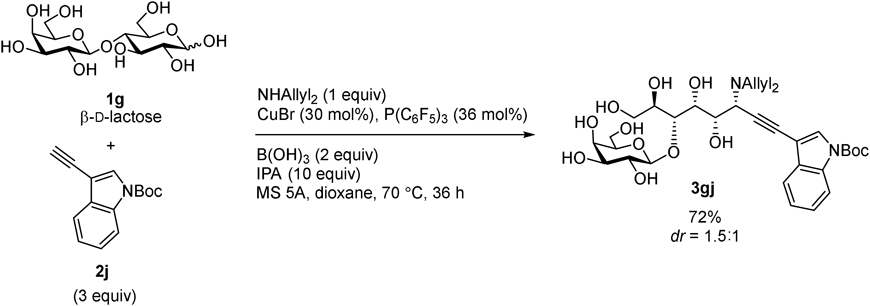
Our first successful entry is aminoalkynylation of unprotected aldoses16) (Figs. 3–5). In situ generated copper acetylide was added to an iminium cation generated through reaction between a secondary amine and aldehyde form of aldoses (Fig. 6). Although the three-component catalytic asymmetric reaction of an amine, a simple aldehyde, and a terminal alkyne has been reported,17) replacement of simple aldehydes to unprotected aldoses was not a simple extension. The major hurdle stems from low concentration of the aldehyde form of aldoses. Addition of an appropriate boron reagent was found essential to promote the C–C bond-forming reaction; reversible complexation between the boron reagent and hydroxy groups18) increases the concentration of the aldehyde form of aldoses, which promotes iminium cation formation and the following alkynylation. The reaction was applicable to various alkynes (2a–k) and aldoses (1a–f) (Fig. 3). The disaccharaide, β-D-lactose (1g), was also transformed to the corresponding product 3gj by slight modification of the reaction conditions: increasing the catalyst loading and elevating the reaction temperature (Fig. 4). The diastereoselectivity, however, was almost 1 : 1 for most of the entries, except for the reactions with D-ribose (1b) and D-lyxose (1d), which exhibited exceptionally high diastereoselecitvity. The results indicate that the stereoselectivity was mainly controlled by the substrate, not by the catalyst. The biotin-conjugated alkyne 2l was also introduced to D-arabinose (1a), which demonstrated the potential utility of the catalysis for the synthesis of chemical tool for biological study (Fig. 5). Thus the soft copper acetylide acts as a carbon nucleophile rather than a Brønsted base in this reaction, and our approach based on soft–hard orthogonal reactivity was demonstrated effective for developing carbon elongation reactions of unprotected aldoses.

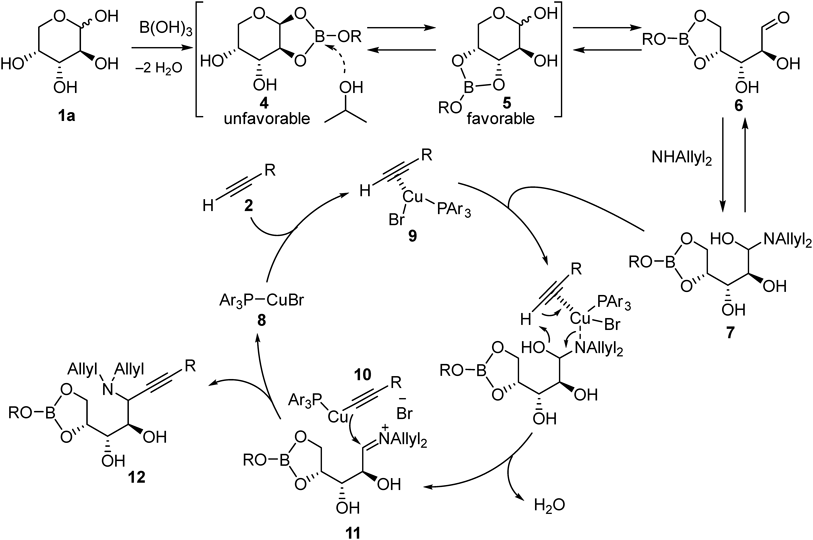
A hypothetical reaction mechanism using D-arabinose (1a) as a representative example is proposed in Fig. 6. Boric acid forms several isomeric complexes with arabinose including an unfavorable complex 4, where aldehyde formation is inhibited. Addition of isopropyl alcohol (IPA) would accelerate the interconversion of carbohydrate–boron complexes, thus promoting preferential formation of a complex 5, where 3,4-cis diol moiety is involved in the complexation. The boron atom in the complex 5 effectively traps the primary hydroxy group, which is generated through ring opening of the hemiacetal structure of complex 5, to stabilize ring-opened structure 6. Following aminoalkynylation of the aldehyde should proceed through the mechanism proposed for the reaction with simple aldehydes.17) A hemiaminal 7 is formed from 6 and diallylamine, which reacts with copper–alkyne complex 9 simultaneously to afford the copper acetylide 10 and the iminium cation 11. Nucleophilic addition of 10 to 11 produces propargylamine 12 and regenerates the catalyst 8.
2.2. Propargylation of Unprotected Aldoses and Sialic Acid SynthesisWe planned synthesis of sialic acid derivatives, which are biologically important 9- or 8-carbon monosaccharides.19) A close analysis of sialic acid derivatives led us to develop propargylation of unprotected aldoses (Fig. 7); after introduction of propargyl group to unprotected aldoses, oxidation of the alkyne moiety to a ketoacid group would furnish sialic acids.
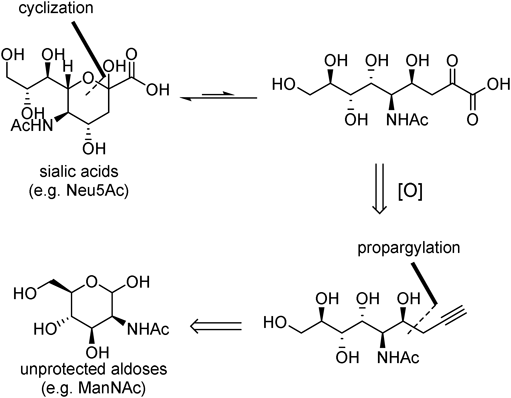
Copper-catalyzed propargylation of unprotected aldoses was realized using allenylboronate 13 as a pro-nucleophile and B(OMe)3 as additive to stabilize the aldehyde form of unprotected aldoses20) (Figs. 8, 9). An important challenge in C–C bond-forming reaction at the anomeric carbon of unprotected aldoses is control of stereochemistry by catalyst. Due to the complex chiral scaffold and multiple hydroxy groups of the substrates, stereochemistry of C–C bond-forming reaction was mostly controlled by the structure of the substrate. Therefore it is critically important that we realized catalyst-controlled propargylation reaction by identifying the large bite angle chiral bisphosphine ligand, Ph-SKP (L1)21): the (S,S,S)-L1/Cu catalyst gave C4-(S) isomer 14 selectively whereas (R,R,R)-L1/Cu catalyst gave C4-(R) isomer 15 selectively. Two different conditions were used depending on the reactivity of the unprotected aldoses. MesCu was used as the copper source in condition A whereas CuClO4(MeCN)4 was used in combination with CF3COOK in condition B. Either condition A or condition B was applicable to a variety of unprotected aldoses ranging from furanoses (1a, 1c, 1d, 1j) and pyranoses (1e, 1h, 1i, 1k, 1l, 1m, 1n, 1o) (Fig. 8) to a disaccharide 1g (Fig. 9), affording 14 or 15 depending on the chirality of the catalyst.

A proposed catalytic cycle is shown in Fig. 10. As in the aminoalkynylation reaction, the concentration of the reactive aldehyde 18 is increased by addition of the boron reagent, B(OMe)3. Copper alkoxide 16, generated by deprotonation of a hydroxy group of the aldose by MesCu, undergoes transmetalation with the allenylboronate 13 to produce allenylcopper 17 as active species. Thus the propargylation reaction proceeds via a rigid six-membered transition state 19 to afford copper alkoxide 20. Finally, transmetalation between 20 and 13 would regenerate allenylcopper species 17. The O–B bonds in 21 are cleaved during the work-up to provide the desired product 14. It is noteworthy that the allenylcopper 17, which is a stronger base than copper acetylide 10, could serve as carbon nucleophile in the presence of multiple free hydroxy groups. Hence the result is in strong support of “soft–hard orthogonal reactivity” approach.

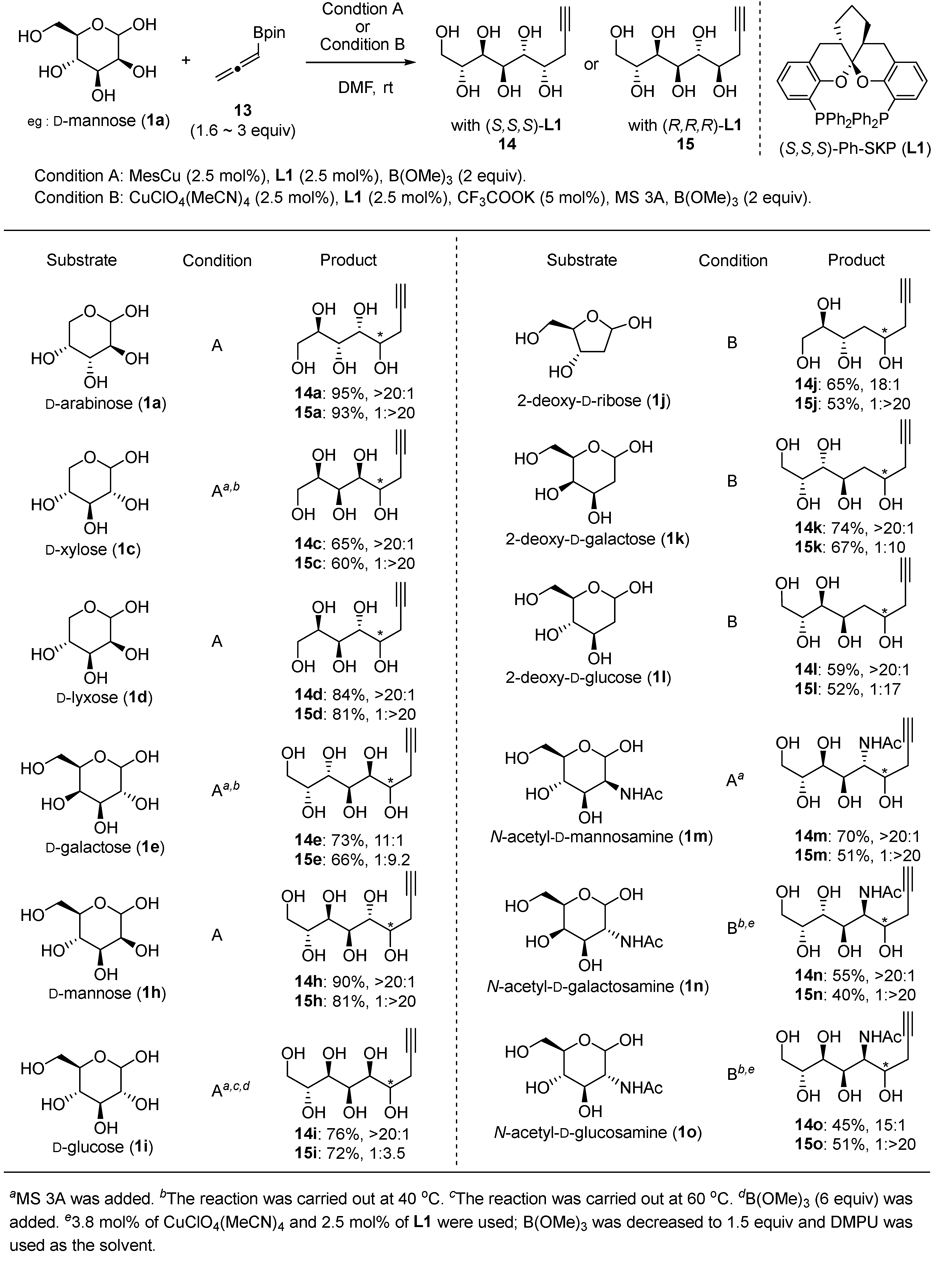
Transformation of propargylation products to sialic acids was successfully developed (Fig. 11). Double oxybromination of the alkyne 14 or 15 afforded a pyrane containing dibromomethyl group 22. The dibromomethyl group was hydrolyzed to its corresponding hydrate followed by oxidation to carboxylic acid to afford sialic acid 23. The protecting-group-minimal four-step transformation, stereodivergent propargylation/oxydibromination/hydrolysis/Pinnick oxidation, was applied to several unprotected aldoses to procure natural (23h, 23m) and unnatural sialic acids (23m′, 23d) including disaccharide derivatives 23g.
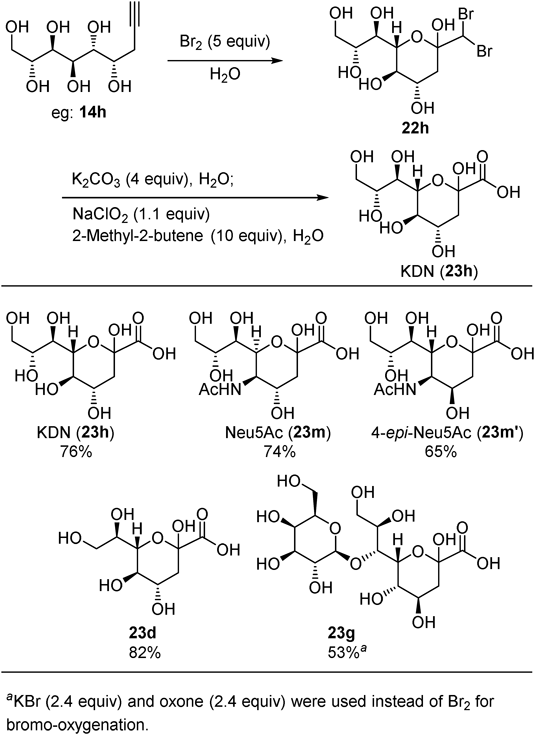
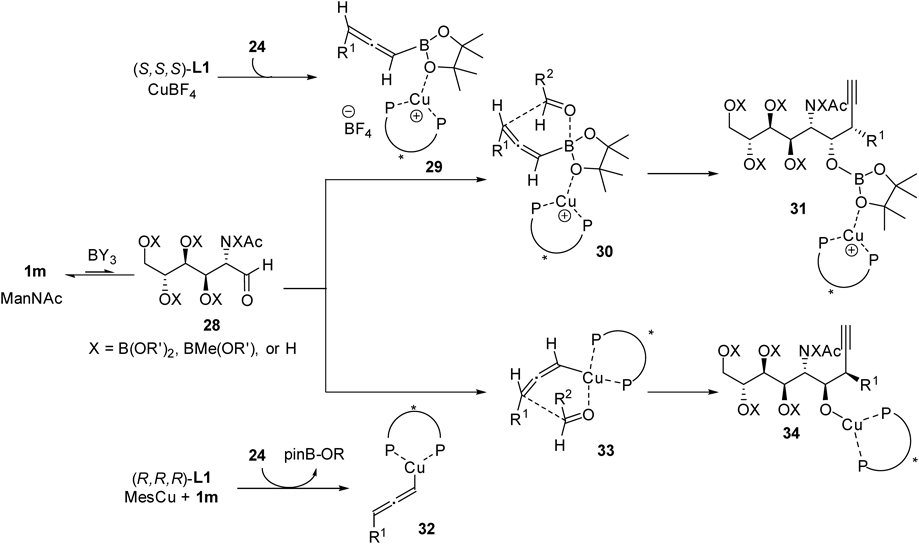
We recently extended the method to C3-substituted sialic acid synthesis using C3-substituted allenylboronates 24 as pro-nucleophiles.22) Although the same reaction conditions described in Fig. 8 were not effective for C3-substituted allenylboronates 24, a close investigation of copper sources and boron additives led to the optimum conditions for both (S,S,S)-L1/Cu catalysis and (R,R,R)- L1/Cu catalysis. Namely, CuBF4 and MeB(OMe)2 for (S,S,S)- L1/Cu catalysis (condition A), MesCu and B(OiPr)3 for (R,R,R)- L1/Cu catalysis (condition B) (Fig. 12). Under these conditions, four allenylboronates (24a–d) were successfully applied to stereodivergent propargylation of D-ManNAc (1m). Notably, sterically demanded cyclohexyl group was introduced with good level of stereoselectivities (25c and 26c).
NMR experiments provided mechanistic insights into the reaction (Fig. 13). Treating the allenylboronate 24a with MesCu (1 equiv) and iPrOH (1 equiv, as a surrogate of aldoses) in DMF-d7 showed generation of protonated allene 27 and iPrOBpin (Figs. 13c, d). On the other hand, no iPrOBpin generation was observed when allenylboronate 24a was treated with CuBF4 (1 equiv) and iPrOH (1 equiv). Instead, a significant upfield shift of Hb (4.9 ppm) to Hb′ (4.5 ppm) and broadened 11B-NMR peak were observed (Figs. 13e, f). The results indicate that the reaction pathway is different between the two conditions (Fig. 14); CuBF4 catalyst acts as a Lewis acid to coordinate to oxygen atom of Bpin group23–25) providing activated allenylboronate 29 (condition A) whereas transmetalation mechanism which generates allenylcopper species 32 along with ROBpin is proposed for MesCu conditions (condition B).26,27) The six-membered transition state in both mechanisms (30 and 33) led to the control of contiguous stereocenters. Thus fine-tuning of the reaction conditions resulted in two different copper catalysts, which promoted highly stereoselective C3-substituted propargylation of unprotected aldoses.

Although the same reaction sequence as mentioned above (Fig. 11) did not provide C3-substituted sialic acids, an alternative protecting-group-minimal synthetic route was developed to afford sialic acids 36b and 38b in two steps from the propargylation products 25b and 26b, respectively (Fig. 15): AgNO3 mediated iodination of terminal alkyne28) followed by OsO4-catalyzed dihydroxylation in the presence of epichlorohydrin to prevent undesired dehydration at the anomeric carbon.29)

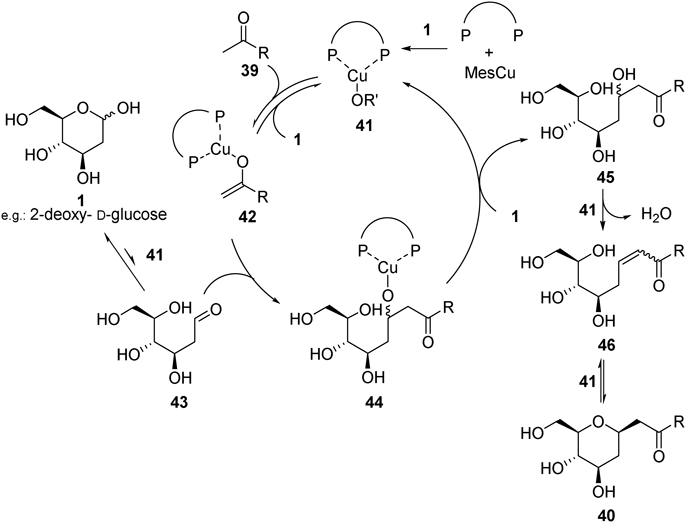
We next developed copper-catalyzed dehydrative ketone incorporation to unprotected aldoses to procure C-glycosides in one operation30) (Fig. 16). The reactivity was correlated to the bite angle of the ligand on copper atom: the ligand of large bite angle gave higher reactivity. The large bite angle ligand might prohibit aggregation of copper(I), which increases the concentration of active monomeric copper(I) species.31,32) Thus the key for the reaction was identification of exceptionally large bite angle ligand L2. Although 2-deoxy-D-ribose (1j) produced a mixture of pyranose-type 40ja and furanose-type 40ja′, other C2-non-substituted aldoses produced pyranose-type C-glycosides (40ka, 40la–40lo, 40pp, 40qa) exclusively in good yields with high β-stereoselectivities at the anomeric carbon. On the other hand, C2-hydroxyaldoses were less reactive and only limited ketones, 4-nitroacetophenone (39e) or 2-pyridylmethyl ketone (39l), were introduced to give 40me (52% yield, α/β = 1: > 20), 40ml (51% yield, α/β = 1 : 5), 40ne (35% yield, α/β = 1 : 7), and 40ge (20% yield, α/β = 1 : 4).

Based on the previous report on ketone incorporation reaction with cyclic hemiaminal,33) a proposed catalytic cycle is shown in Fig. 17. The concentration of aldehyde form of aldoses 43 would be slightly increased by the basic copper alkoxide 41, which is generated through reaction between MesCu and unprotected aldoses. The copper enolate 42, generated through deprotonation of α-proton of the ketone 39 by copper alkoxide 41, preferentially reacts with aldehyde form of the aldoses 43, rather than protonolysis by free hydroxy groups, to give copper aldolate 44. Protonation of 44 regenerates copper alkoxide 41 and produces aldol product 45. Dehydration from 45 and following intramolecular conjugate addition of the hydroxy group of 46 are also promoted by copper alkoxide 41 to provide 40. Note that addition of boron compounds is not required in this reaction probably due to high nucleophilicity of the copper enolate.
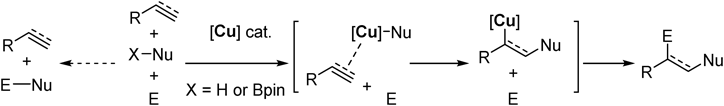
C–C multiple bonds are ubiquitous and stable functional group in feedstock molecules as well as functional molecules. Addition of two distinct functional groups on each side of C–C multiple bonds in a single operation, namely difunctionalization, is a highly attractive transformation that can rapidly elevate molecular complexity.34–37) Difunctionalization, however, involves two reactive reagents along with the C–C multiple bond. Hence precise control of reaction order, that is to say chemoselectivity, is necessary to avoid undesired direct reaction between the two reagents without involving the C–C multiple bond (Fig. 18). We expected that a “soft” nucleophilic copper species preferentially reacts with “soft” C–C multiple bonds to circumvent undesired reaction pathway, realizing difunctionalization.
To assess our working hypothesis, we first investigated oxycupration of allenes followed by asymmetric addition to carbonyl compounds using substrate 47, which contains a hydroxy group and an allene moiety in one molecule38) (Fig. 19). The designed substrates facilitated control of reaction order by making the first step intramolecular reaction. (R)-DTBM-SEGPHOS (L3)/MesCu catalyst was used with Al(OtBu)3 co-catalyst for the substrates containing primary alcohols (47a–d) (condition A), and (S,S)-Ph-BPE (L4)/MesCu catalyst was used without Al(OtBu)3 co-catalyst for the substrates containing tertiary alcohols (47e–h) (condition B). Addition of Al(OtBu)3 co-catalyst improved product yield, especially in the reaction with aliphatic aldehydes. The reaction proceeded in high enantioselectivity (81–95% enantiomeric excess (ee)) with various aromatic (48a, 48h–n) and aliphatic aldehydes (48b–f). A ketone 48g was also applicable albeit with moderate enantioselectivity (49ag: 77% yield, 76% ee). A notable example is a reaction with the primary alcohol containing aldehyde 48n, which afforded 49en (79% yield, 89% ee). The reaction proceeded smoothly without protonolysis of the in situ generated allylcopper species by free hydroxy group, further supporting the “soft–hard orthogonal reactivity” concept.

We propose a catalytic cycle as shown in Fig. 20. First, copper alkoxide 50, generated through deprotonation of the hydroxy group by the catalyst, undergoes oxycupration of the allene to afford allylcopper 52 and/or 53. Addition of the thus-generated allylcopper to aldehyde 48 proceeds through a six-membered transition state 55 to give copper alkoxide 56. Ligand exchange on the copper atom produces 49 with regeneration of active species 50. Because the enantioselectivity was not affected by addition of Al(OtBu)3, the additive would contribute to acceleration of the ligand exchange step after the enantioselectivity was determined.

We extended this strategy to formation of chiral 2-(2-hydroxyethyl)indole derivatives using allenylanilides as substrates39) (Fig. 21). Addition of Mg(OiPr)2 co-catalyst instead of Al(OtBu)3 effectively increased the yield of reactions with aliphatic aldehydes and ketones.

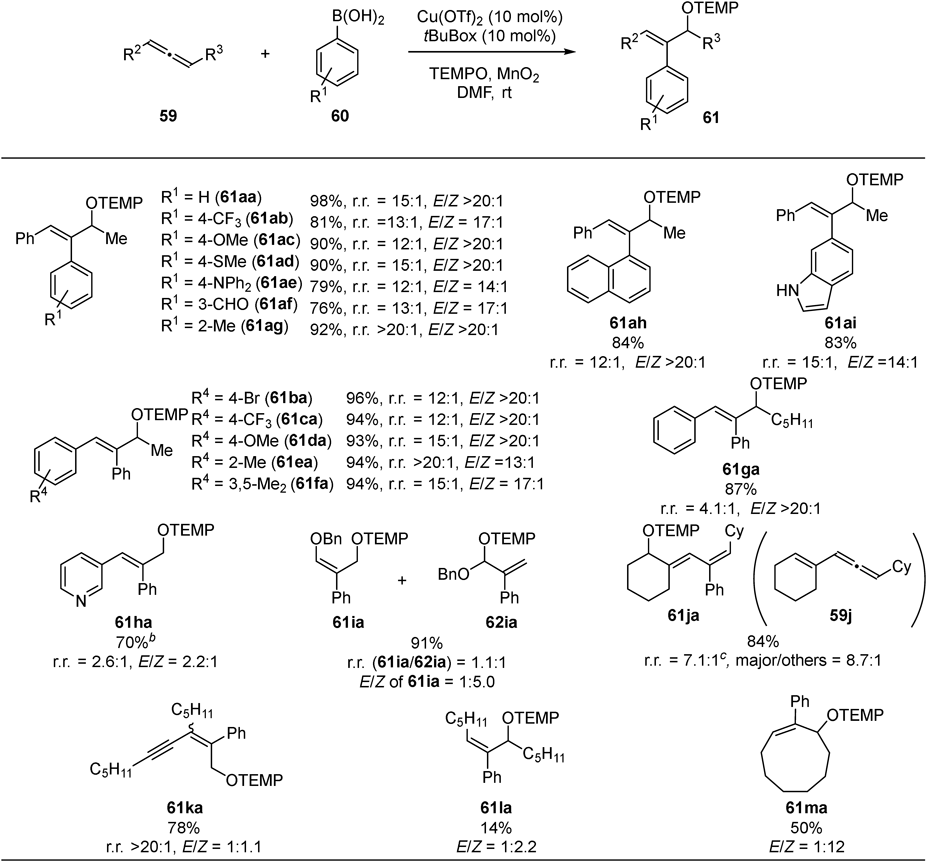
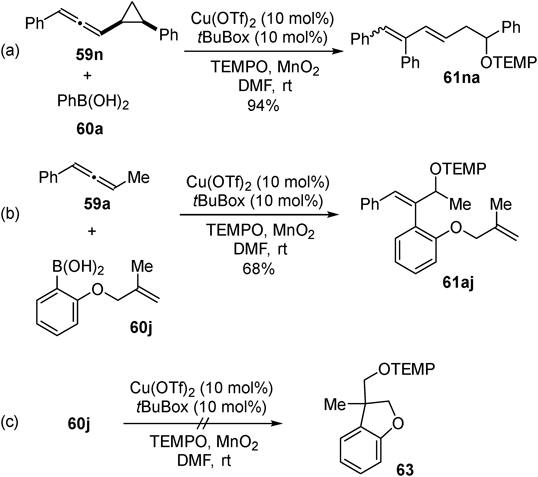
We subsequently developed an intermolecular three-component reaction: oxyarylation of allenes using arylboronic acids as carbon source and 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) as oxygen source40) (Fig. 22). Reaction of arylallenes (59a–g) proceeded with high regioselectivity using various arylboronic acids (60a–i). Functional group tolerance is also good, highlighted by the compatibility of highly reactive formyl group (61af: 76% yield, r.r. = 13 : 1, E/Z = 17 : 1). Other types of allenes including pyridyl allene 59h, benzyloxy allene 59i, and vinyl allene 59j were also reactive substrates to give the products (61ha, 61ia + 62ia, 61ja), albeit with lower regioselectivities. Alkynyl allene 59k was competent to give the product 61ka with high regioselectivity (78% yield, r.r. >20 : 1, E/Z = 1 : 1). On the other hand, alkyl allene 59l exhibited low reactivity (61la: 14% yield) whereas strained cyclic allene 59m gave moderate yield (61ma: 50% yield).
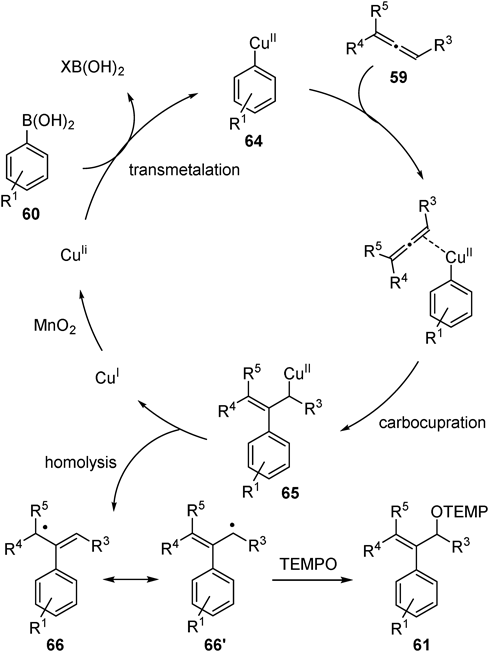
A radical clock experiment using allene 59n containing cyclopropane ring afforded ring-opened product 61na exclusively (Fig. 23a). In addition, reaction with boronic acid 60j provided product 61aj (Fig. 23b) whereas cyclized product 63 was not detected even in the absence of allenes (Fig. 23c). The results indicate no aryl radical generation under the reaction conditions.41) A proposed reaction mechanism based on these observations is as follows (Fig. 24). First, transmetalation between the copper catalyst and arylboronic acid 60 generates arylcopper(II) species 64. Carbocupration of allenes proceeds to afford allylcopper(II) 65, and following homolysis of the C–Cu(II) bond42) generates allyl radical species 66 and 66′. Finally, the allyl radical was trapped by a TEMPO to give the oxyarylation product 61 and oxidation of Cu(I) by MnO2 regenerates Cu(II).
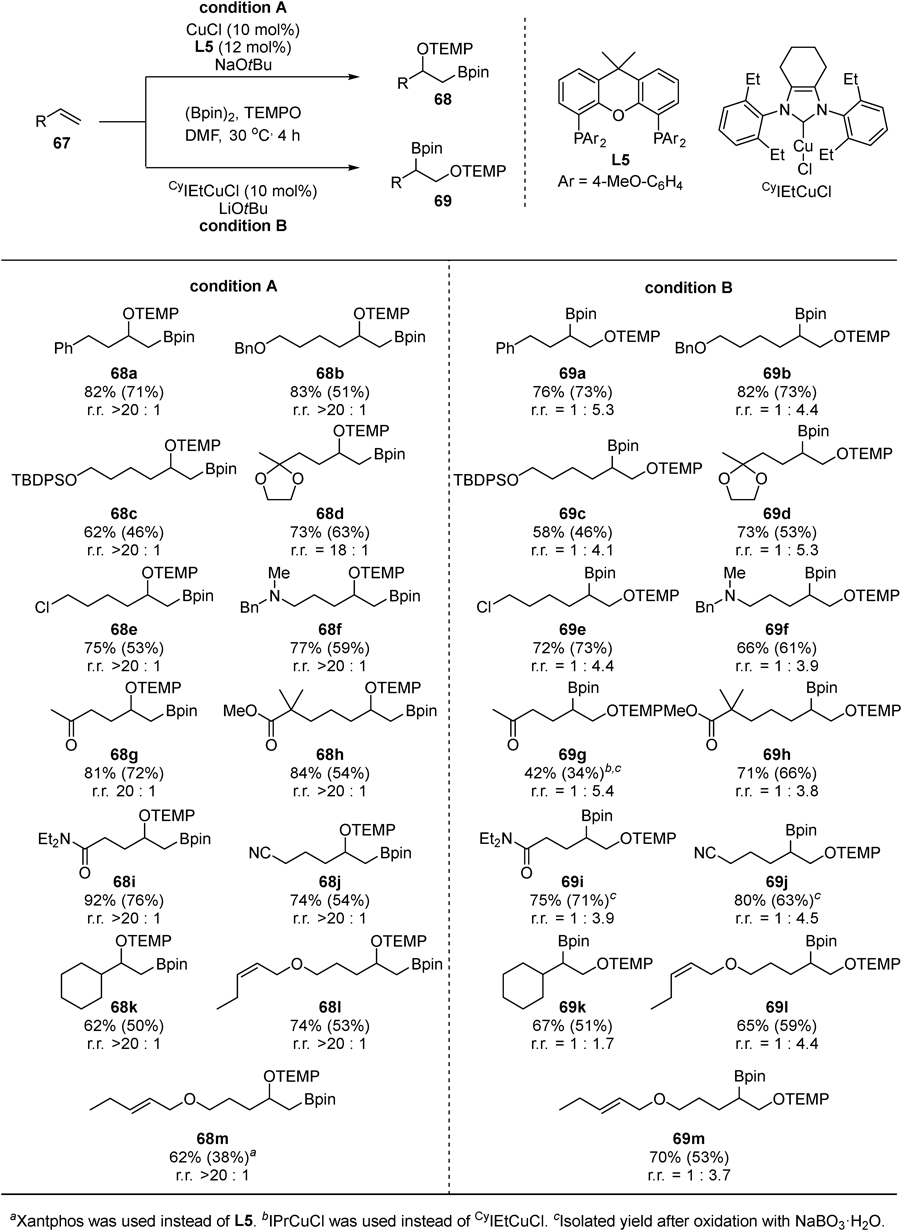
Next, we tackled to develop difunctionalization reactions of alkenes, which are generally less reactive than allenes due to lower polarizability. Based on the knowledge obtained by oxyarylation of allenes and the high reactivity of borylcopper species toward alkenes,43) we developed oxyboration of unactivated alkenes using (Bpin)2 as boron source and TEMPO as oxygen source44) (Fig. 25). The reaction proceeded with two types of copper catalysts and each provided different regioselectivity: β-oxygenated primary alkylboronates 68 were obtained with 4′-MeO-Xantphos (L5)/CuCl (condition A) whereas β-oxygenated secondary alkylboronates 69 were obtained with CyIEtCuCl (condition B). The copper catalyst chemoselectively reacted at the terminal alkene in the presence of other functional groups, such as ethers (68b–d and 69b–d), an amine (68f and 69f), carbonyl groups (68g–i and 69g–i), and even internal alkenes (68l, m and 69l, m).
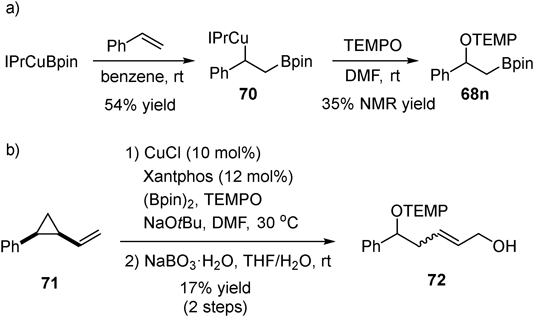
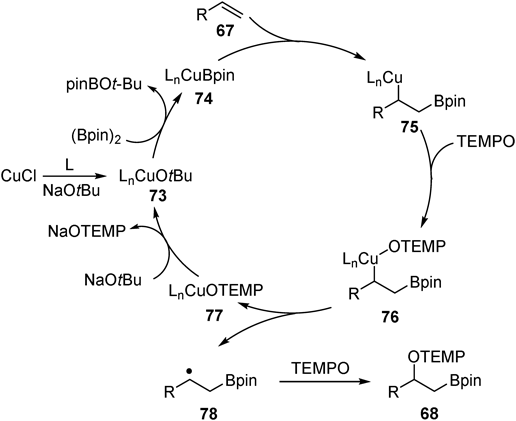
After borylcupration of styrene, plausible alkylcopper species 70 was isolated45) and reacted with TEMPO to give oxyboration product 68n (Fig. 26a). Additionally, radical clock experiment using vinylcyclopropane 71 produced ring-opened product 72 in 17% yield (Fig. 26b). These results suggest that oxygenation of C–Cu bond by TEMPO proceeds through radical intermediate. A plausible catalytic cycle is as follows (Fig. 27). Borylcupration proceeds with alkenes via in situ generated nucleophilic borylcopper species 74 to afford alkylcopper(I) intermediate 75. The alkylcopper(I) 75 was oxidized by TEMPO to alkylcopper(II) 76. The labile C–Cu(II) bond was homolytically cleaved to generate alkyl radical 78. Finally, the alkyl radical 78 was trapped by TEMPO to afford oxyboration product 68. A combination of two distinct properties of the copper-containing intermediates, namely the nucleophilic property of borylcopper species in the borylcupration of alkenes and the one-electron redox-active property of alkylcopper species in the generation of alkyl radical, was key to success.
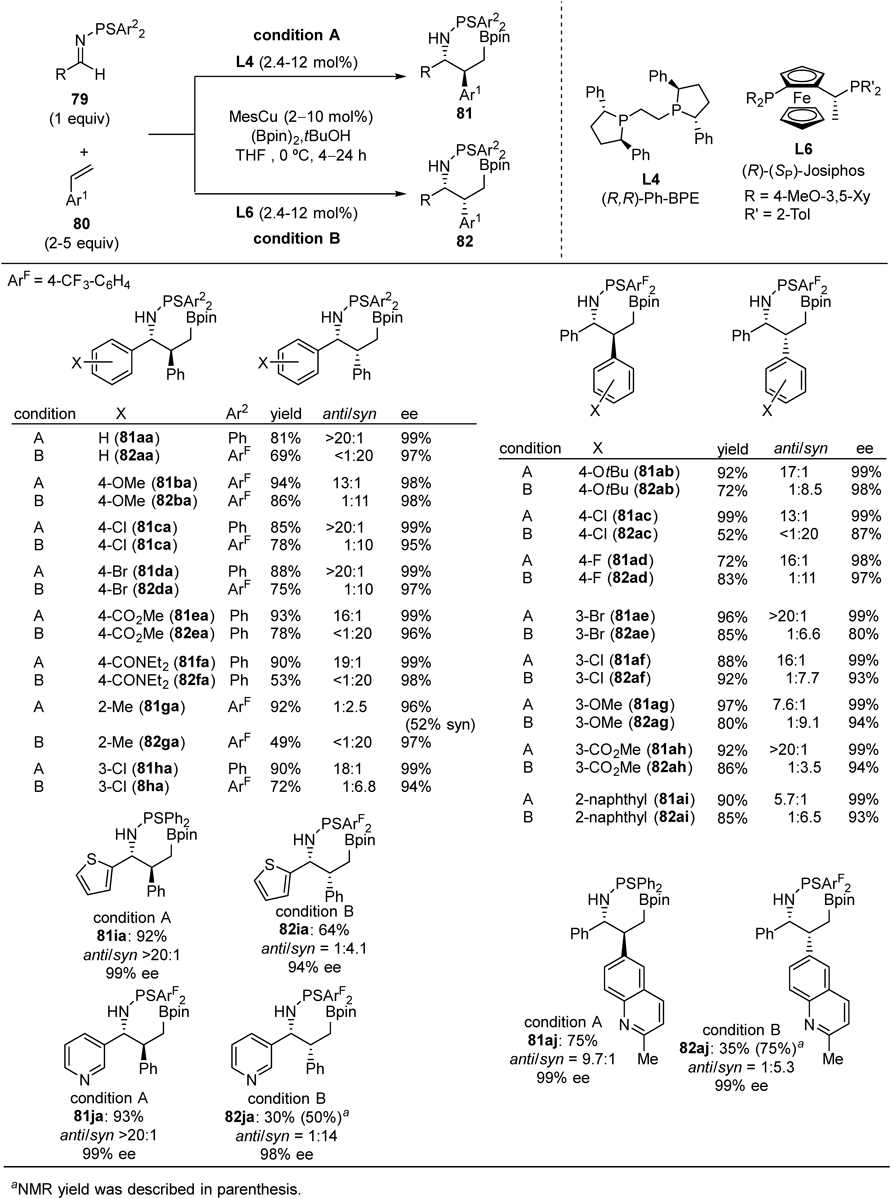
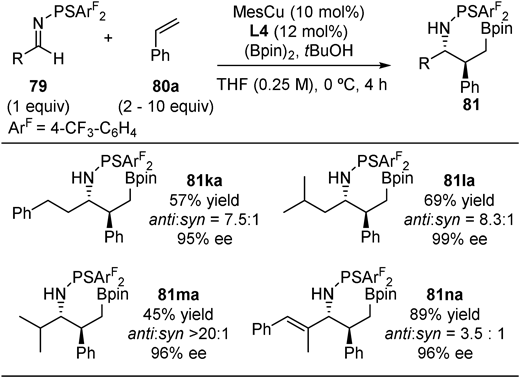
The other entry of difunctionalization of alkenes is borylative coupling of styrenes and thiophosphinoylimines46) (Fig. 28). The diastereoselectivity as well as the enantioselectivity could be controlled by chiral ligands on the copper atom: both anti- and syn-diastereomers were obtained with high enantioselectivities by selecting either L4 or L6 as a chiral ligand. Various aryl imines (79a–j) and styrene derivatives including heteroarene derivatives (80a–j) were applicable, giving both diastereomers (81 and 82) in high enantioselectivities and diastereoselectivities. Application of alkyl imines (79k–n) was possible with L4/MesCu catalysis to afford anti-product (81ka–81na) whereas L6/MesCu catalysis did not work in this case (Fig. 29).
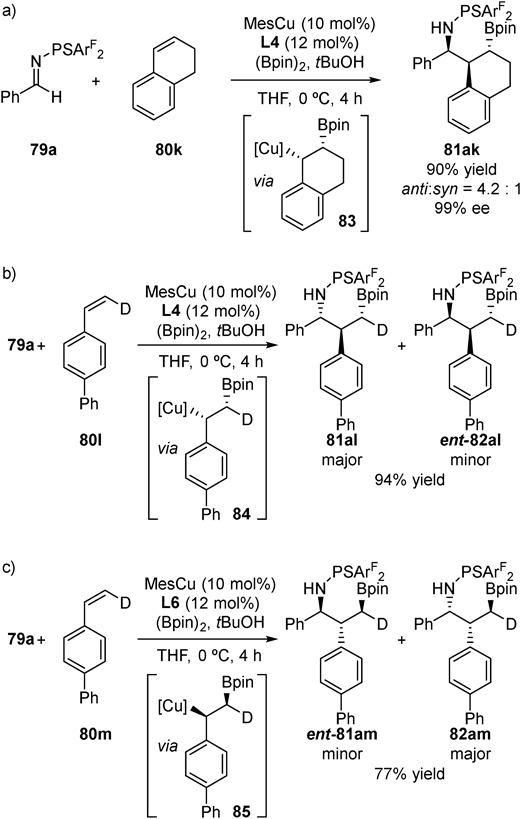
An experiment using cyclic styrene derivative 80k revealed that the C–C bond is formed from the opposite face of the C–B bond (Fig. 30a). Because borylcupration is known to proceed with syn-selectivity,48) the observation indicated that the C–C bond formation occurred from the backside of C–Cu bond via 83. This backside attack process was further supported by two experiments (Figs. 30b, c) using deuterated styrene derivatives (80l and 80m) under either L4/MesCu conditions or L6/MesCu conditions, giving stereo-inverted products at benzylic position from the plausible benzylic copper intermediates (84 and 85). Based on these observations and previous reports, we propose following catalytic cycle (Fig. 31). After borylcupration of styrene, addition of the generated benzylcopper species 86 to imine 79 proceeds from backside of C–Cu bond. This addition step proceeds through a flexible linear transition state 87, which can be controlled by steric effect of copper catalyst, leading to chiral ligand-dependent stereoselectivity. Thus-generated copper amide intermediate 88 is protonated by tBuOH to afford the three-component coupling product 81 and regenerate CuOtBu.

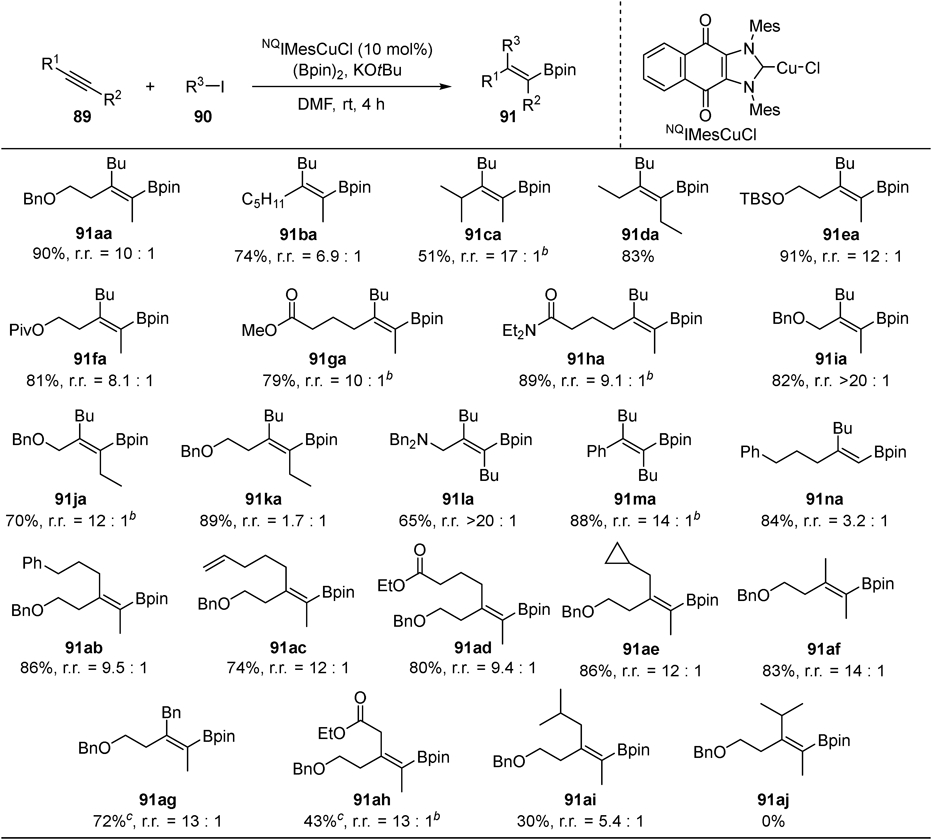
We also succeeded in difunctionalization of alkynes (Fig. 32): regioselective and stereoselective alkylboration of internal alkynes 89.48) Although copper-catalyzed alkylboration of alkynes has been previously reported by other groups,49–54) significant expansion of substrate scope of alkynes 89 and alkylating agents 90 was realized by identifying naphthoquinone-conjugated NHC55) copper catalyst (NQIMesCuCl). When other ligands were used, the reaction between in situ generated borylcopper species and alkylhalides without incorporating alkenes occurred predominantly. The π-accepting naphthoquinone-conjugated NHC copper catalyst would exhibit increased affinity toward the π-electrons of alkynes, resulting in chemoselective promotion of the borylcupration of alkynes, rather than undesired borylation of alkylhalides. Functional group compatibility is good: carbonyl groups (91ga, 91ha, 91ad, 91ah) and an alkenyl moiety (91ac) were intact under the conditions.
In summary, we developed chemoselective reactions by exploiting the “soft” property of copper catalyst: soft–hard orthogonal reactivity between soft copper catalyst and hard hydroxy group, and soft–soft preferential reactivity between copper catalysts and C–C multiple bonds. These reactions enable unprecedented transformations that would contribute to streamlining synthesis of drug lead compounds and facilitate late-stage functionalization of poly-functionalized pharmaceuticals and natural products.
I would like to express my sincere gratitude to Prof. Motomu Kanai (The University of Tokyo) for generous support, encouragement and fruitful advice to achieve the results described in this review. I am also grateful to all my coworkers who dedicated their tremendous efforts on this research. I appreciate thoughtful advice of Prof. Masaya Sawamura on writing this review. The work described herein was supported by a Grant-in-Aid for Young Scientists (B), and Scientific Research (C) from the Japan Society for the Promotion of Science.
The author declares no conflict of interest.
This review of the author’s work was written by the author upon receiving the 2019 Pharmaceutical Society of Japan Award for Young Scientists.