Abstract
Rathke’s cleft cyst (RCC) is a common incidental tumor in the hypothalamic-pituitary region. Some reports have shown that the clinical symptoms and endocrine functions of symptomatic RCCs are temporarily improved by glucocorticoid administration. However, it is still unknown whether glucocorticoid treatment is effective for symptomatic RCCs according to long-term observations. In this study, we describe the long-term clinical outcomes of two cases of glucocorticoid-treated biopsy-proven secondary hypophysitis caused by RCCs. We summarize the symptoms, imaging findings, and endocrine evaluations of two symptomatic RCC patients with concomitant hypophysitis before and after prednisolone treatment. In both evaluated cases, visual impairments and altered endocrine parameters were present due to chiasm and stalk compression; these outcomes improved after shrinkage of RCCs in response to prednisolone administration, and partial recovery of anterior pituitary hormone secretion was observed. However, in both cases, the deficits in anterior pituitary hormone secretion recurred, possibly due to persistent inflammatory infiltration in the RCCs and pituitary glands. After relapse of hypophysitis, anterior hormone secretion did not fully recover. In our cases of secondary hypophysitis caused by RCCs, prednisolone administration had an early effect of cyst shrinkage, followed by partial improvements in clinical symptoms and pituitary functions. However, long-term observation showed that prednisolone treatment did not contribute to complete improvement in anterior pituitary hormone dysfunction.
RATHKE’S CLEFT CYST (RCC) is a common disorder of the hypothalamic-pituitary region. In terms of embryology, the anterior and intermediate lobes of the pituitary gland arise from the so-called Rathke’s pouch. RCCs are inadequate remnants found on autopsy in the pituitary glands of approximately 4% to 20% of adults [1, 2]. Most RCCs are asymptomatic, but some are symptomatic, accompanied by headaches, visual disturbances, and pituitary hormone dysfunction. Symptomatic RCCs are mainly caused by compression during cyst expansion and/or by secondary hypophysitis due to inflammatory changes triggered by spreading of the liquid content from collapsed cysts to the pituitary gland and adjacent tissues [3-5].
Generally, surgical drainage with partial excision of the cyst wall is considered for symptomatic RCCs to improve headache, visual dysfunction, and endocrine disorders [6]. However, most preoperative endocrine dysfunctions, including diabetes insipidus (DI), are usually irreversible and worsen after surgery. In fact, it has been reported that only 14% to 18% of patients show improvements in endocrine function, and 19% of patients acquire DI after surgical treatment, with an overall five-year recurrence rate of 18% [7]. Such effects of the treatment, clinical course, and features of RCC have been reported by several authors [8-13].
Recently, glucocorticoids have been reported as an effective treatment for RCC [14]. The anti-inflammatory effect of glucocorticoids may be partially responsible for short-term alterations in cyst size and can be considered for symptomatic RCCs with inflammation. However, the efficacy of glucocorticoids, more specifically prednisolone, on clinical symptoms and endocrine functions in RCC patients remains unclear. In this report, we describe the outcomes of prednisolone treatment in two cases of biopsy-proven symptomatic RCC with secondary hypophysitis.
In both of our cases, prednisolone administration had an early effect of cyst shrinkage, followed by partial improvements in clinical symptoms and pituitary functions. However, long-term observations showed that prednisolone treatment did not maintain the initial improvement in anterior pituitary hormone secretion, resulting in permanent endocrine dysfunction.
Case Reports
Case 1
A 60-year-old Asian woman with a history of dyslipidemia presented to our hospital with general fatigue, polydipsia, polyuria, asitia and nausea. She had no history of hemorrhage or trauma and did not have visual deficits.
T1-weighted magnetic resonance imaging (MRI) with heterogeneous gadolinium enhancement revealed swelling of the pituitary gland and stalk with an asymmetrical multicystic appearance (Fig. 1). Pharmacologic loading tests revealed an excessive response of ACTH to CRH (baseline ACTH <5.0 pg/mL, 30-min peak ACTH 181 pg/mL) and no response on the insulin tolerance test (ITT; baseline ACTH <5.0 pg/mL, peak ACTH 5.0 pg/mL) (Table 1). Laboratory findings also showed secondary hypogonadism, secondary hypothyroidism and hyperprolactinemia (Table 1). Furthermore, GH showed a low response to the ITT, fulfilling the criteria for adult growth hormone deficiency (AGHD) (Table 1). Therefore, the patient was diagnosed with combined pituitary hormone deficiency. Moreover, a hypertonic saline test confirmed the diagnosis of central diabetes insipidus (DI). Based on the radiologic findings, these hormonal deficiencies were believed to have been caused by lymphocytic hypophysitis.
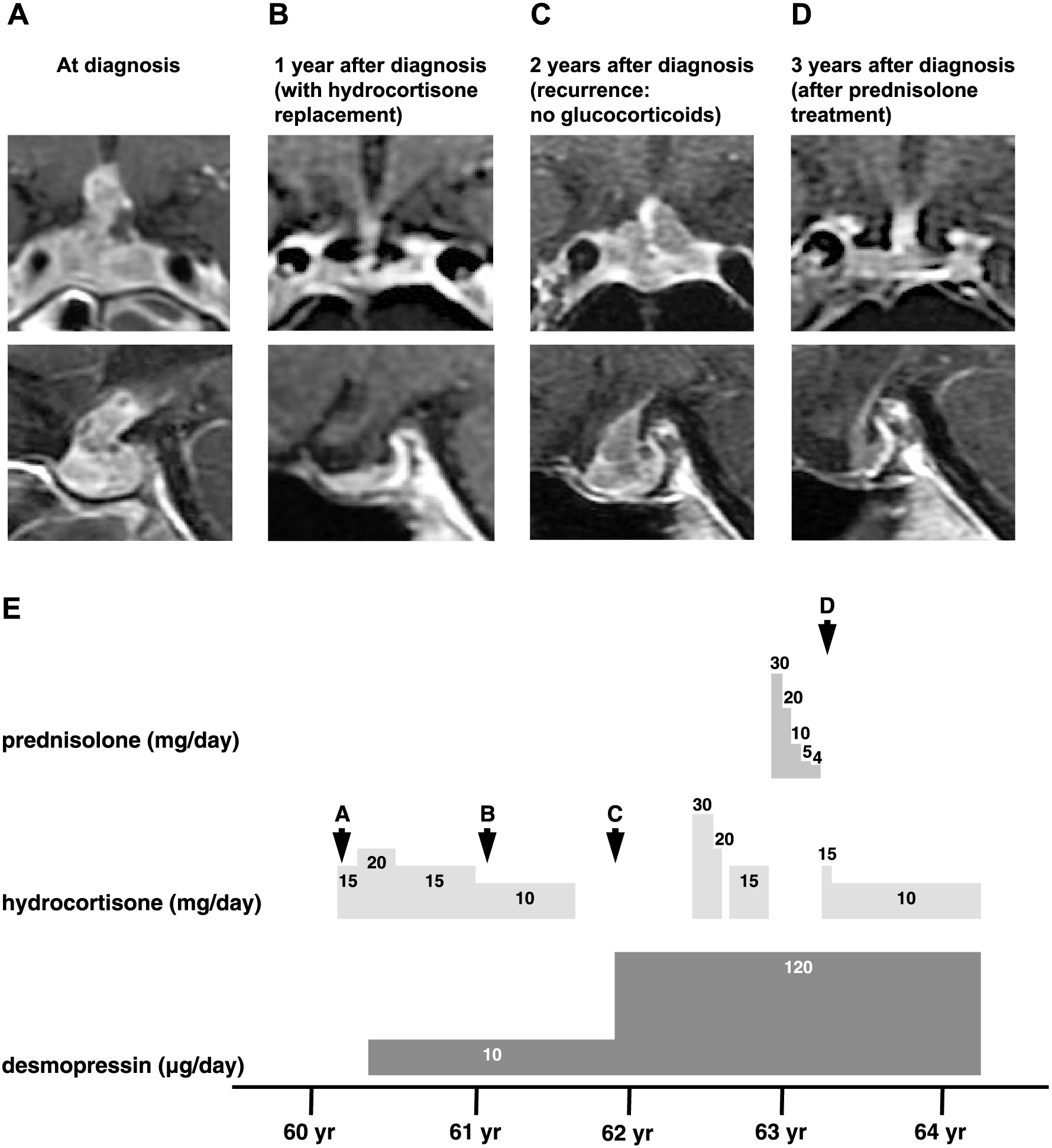
Table 1
Summary of the results of loading tests in Case 1
|
Time after diagnosis |
At diagnosis |
1.5 years |
2 years |
3 years |
6 years |
| Glucocorticoids |
Hydrocortisone 15 mg/day p.o. |
Hydrocortisone 10 mg/day p.o. |
No glucocorticoids |
Hydrocortisone 10 mg/day p.o. (1 month after PSL treatment: total PSL 1,962 mg p.o.) |
Hydrocortisone 10 mg/day p.o. (2.5 years after PSL treatment: total PSL 1,962 mg p.o.) |
| Time after bioposy |
— |
— |
10 days |
1 year |
3.5 years |
| CAPST |
Normal range |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
| ACTH (pg/mL) |
<46.0 |
<5.0 |
181.0 |
21.3 |
133.0 |
9.2 |
21.3 |
10.5 |
16.8 |
8.1 |
8.1 |
| Cortisol (μg/dL) |
5.0–25.0 |
5.5 |
26.8 |
8.9 |
20.7 |
3.2 |
6.8 |
3.0 |
5.6 |
<1.0 |
<1.0 |
| GH (ng/mL) |
0.04–3.60 |
1.58 |
15.30 |
0.77 |
9.31 |
0.17 |
4.54 |
0.11 |
3.08 |
0.35 |
3.73 |
| TSH (μIU/mL) |
0.35–4.94 |
0.701 |
5.213 |
3.134 |
28.250 |
2.807 |
10.715 |
2.608 |
27.939 |
0.091 |
0.498 |
| PRL (pg/mL) |
5.18–26.53 |
112.70 |
183.68 |
11.47 |
35.21 |
14.15 |
41.60 |
22.09 |
55.68 |
24.02 |
30.87 |
| LH (mIU/mL) |
1.76–10.24 |
0.02 |
0.59 |
0.03 |
0.68 |
0.02 |
0.38 |
0.01 |
0.17 |
0.02 |
0.09 |
| FSH (mIU/mL) |
3.21–14.72 |
0.45 |
3.66 |
0.47 |
3.27 |
0.39 |
1.93 |
0.17 |
1.16 |
0.11 |
0.67 |
| E2 (pg/mL) |
|
<10 |
N.A. |
<10 |
N.A. |
<10 |
N.A. |
<10 |
N.A. |
<10 |
N.A. |
| FT4 (ng/dL) |
0.70–1.48 |
0.82 |
N.A. |
0.97 |
N.A. |
0.66 |
N.A. |
0.81 |
N.A. |
0.89 |
N.A. |
| IGF-1 (ng/mL) |
147–358 |
50.1 |
N.A. |
69 |
N.A. |
77 |
N.A. |
29 |
N.A. |
25 |
N.A. |
| ITT |
Normal range |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
| ACTH (pg/mL) |
<46.0 |
<5.0 |
5.0 |
11.4 |
112.0 |
8.3 |
32.2 |
9.1 |
16.7 |
8.7 |
10.2 |
| Cortisol (μg/dL) |
5.0–25.0 |
1.9 |
1.7 |
6.5 |
18.9 |
2.8 |
10.1 |
9.5 |
16.4 |
<1.0 |
<1.0 |
| GH (ng/mL) |
0.04–3.60 |
2.80 |
2.62 |
0.33 |
1.04 |
0.27 |
0.26 |
0.11 |
0.13 |
0.33 |
0.30 |
| GHRP-2 loading test |
Normal range |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
| GH (ng/mL) |
0.04–3.60 |
0.82 |
6.26 |
0.90 |
11.70 |
0.18 |
1.52 |
0.14 |
1.73 |
0.19 |
0.91 |
| ACTH (pg/mL) |
<46.0 |
<5.0 |
65.0 |
21.5 |
35.0 |
9.8 |
15.3 |
10.3 |
16.3 |
5.2 |
11.8 |
| Cortisol (μg/dL) |
5.0–25.0 |
8.1 |
20.5 |
12.4 |
15.0 |
4.5 |
6.7 |
6.5 |
9.7 |
<1.0 |
<1.0 |
Abbreviations: CAPST, combined anterior pituitary stimulation test (100 μg CRH, 100 μg GRH, 500 μg TRH, and 100 μg LHRH); ITT, insulin tolerance test; GHRP-2, growth hormone-related peptide-2; PSL, prednisolone; N.A., Not Available; p.o., per os
Since infectious diseases, such as tuberculosis, were not completely ruled out, we initially avoided prednisolone and treated the patient with a relatively high dosage of hydrocortisone (30 mg/day). The dosage of hydrocortisone was gradually reduced from 30 mg/day to 10 mg/day over one and a half years, with a total dose of 6,365 mg per os (Fig. 1). After one and a half years of hydrocortisone treatment, the size of the cyst regressed (Fig. 1), and the baseline ACTH, cortisol, TSH, and PRL levels recovered (Table 1). Additionally, the response of ACTH to the ITT recovered (baseline ACTH 11.4 pg/mL, peak ACTH 112.0 pg/mL), as well as the responses of PRL and TSH to TRH (Table 1). Taking into account this recovery of adrenal cortical function, hydrocortisone was terminated. However, after complete discontinuation of hydrocortisone, re-enlargement of the cyst was observed (Fig. 1). To confirm the diagnosis, we performed a transsphenoidal pituitary biopsy. The pathological findings showed ciliated columnar cells with extensive inflammation of the cyst capsule extending into the hypophyseal tissue as well as squamous metaplasia of the cyst wall (Fig. 2), the findings of which were compatible with RCCs. The inflammatory reaction consisted mainly of lymphocytes with numerous plasma cells, and a few multinucleated macrophages were detected amid the inflammatory cells (Fig. 2), which led to a diagnosis of hypophysitis secondary to RCC. The enlarged pituitary gland slightly shrank after biopsy, and the loading tests performed 10 days after biopsy showed decreases in baseline ACTH, cortisol, and TSH levels, as well as the ACTH response to CRH (baseline ACTH 9.2 pg/mL, 60-min peak ACTH 21.3 pg/mL) and the ITT (baseline ACTH 8.3 pg/mL, peak ACTH 32.2 pg/mL) (Table 1).

We then treated the patient with prednisolone (initial dosage 30 mg/day). The dosage of prednisolone was gradually reduced over the course of 5 months, with a total dose of 1,962 mg per os, and was switched to hydrocortisone (15 mg/day) (Fig. 1). Although the cyst volume further decreased after prednisolone loading (Fig. 1), the loading tests performed 1 month after prednisolone treatment showed low responses of ACTH, cortisol, GH, LH, and FSH to their respective loading tests (CRH stimulation test: baseline ACTH 10.5 pg/mL, 120-min peak ACTH 16.8 pg/mL, ITT: baseline ACTH 9.1 pg/mL, peak ACTH 16.7 pg/mL) (Table 1). The low response of ACTH and cortisol could be due to prednisolone loading since the evaluations were performed immediately after prednisolone treatment. To assess the endocrinological state of this patient without the effect of prednisolone, we re-evaluated the pharmacological tests 2.5 years after prednisolone treatment. The baseline ACTH and cortisol levels were lower than those 1 month after prednisolone treatment. ACTH showed no response to CRH (baseline ACTH 8.1 pg/mL, 15-min peak ACTH 8.1 pg/mL) and a low response to the ITT (baseline ACTH 8.7 pg/mL, peak ACTH 10.2 pg/mL), but the response to GHRP-2 remained (baseline ACTH 5.2 pg/mL, peak ACTH 11.8 pg/mL) (Table 1). On the other hand, cortisol showed no response to CRH, the ITT, or GHRP-2 (Table 1).
Case 2
A 49-year-old Asian female complained of general fatigue. She was diagnosed with hypophysitis when she was 35 years old and was treated with prednisolone at the former hospital. She had amenorrhea, polydipsia and polyuria, and T1-weighted MRI of the brain showed swelling of the pituitary gland and stalk (Fig. 3). Her endocrinological evaluation revealed an excessive response of ACTH to CRH (baseline ACTH 44 pg/mL, 30-min peak ACTH 306 pg/mL) and a normal response to the ITT (baseline ACTH 26.0 pg/mL, peak ACTH 81.0 pg/mL) (Table 2). TSH, PRL, LH, FSH, and GH showed normal responses to their respective loading tests (Table 2). Moreover, the water deprivation test and desmopressin loading test confirmed the diagnosis of central DI. No pathological analysis was conducted. After prednisolone treatment, there was a slight regression of swelling in the pituitary gland and stalk (Fig. 3); additionally, ACTH no longer excessively responded to CRH (baseline ACTH 36 pg/mL, 15-min peak ACTH 62 pg/mL) but excessively responded to the ITT (baseline ACTH 28 pg/mL, peak ACTH 257 pg/mL) (Table 2). Hypogonadism and DI remained even after prednisolone treatment. She was treated with Kaufman therapy (cyclic administration of estradiol and hydroxyprogesterone) and desmopressin.
Table 2
Summary of the results of loading tests in Case 2
|
Time after diagnosis |
At diagnosis |
0.5 years |
15 years |
15.5 years |
16 years |
18 years |
| Glucocorticoids |
No gulcocorticoids |
No gulcocorticoids (6 months after 1st PSL treatment:
total PSL N.A.) |
No glucocorticoids |
Hydrocortisone 20 mg/day p.o. |
Hydrocortisone 10 mg/day p.o. (1 month after 2nd PSL
treatment: total PSL 1,621 mg p.o.) |
Hydrocortisone 15 mg/day p.o. (2.5 years after 2nd
PSL treatment: total PSL 1,621 mg p.o.) |
| Time after bioposy |
— |
— |
— |
2 months |
7 months |
3 years |
| CAPST |
Normal range |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
| ACTH (pg/mL) |
<46.0 |
44 |
306 |
36 |
62 |
14.9 |
203.0 |
<5.0 |
49.7 |
<5.0 |
21.9 |
<5.0 |
55.7 |
| Cortisol (μg/dL) |
5.0–25.0 |
17.9 |
26.1 |
15.2 |
18.1 |
3.0 |
15.2 |
<1.0 |
1.5 |
<1.0 |
<1.0 |
<1.0 |
<1.0 |
| GH (ng/mL) |
0.04–3.60 |
0.94 |
13.0 |
0.52 |
28.2 |
0.26 |
20.4 |
0.27 |
9.85 |
0.05 |
4.24 |
0.44 |
3.79 |
| TSH (μIU/mL) |
0.35–4.94 |
1.96 |
29.8 |
2.83 |
25.0 |
3.641 |
40.990 |
0.105 |
0.703 |
0.179 |
1.530 |
0.033 |
0.277 |
| PRL (pg/mL) |
5.18–26.53 |
22 |
52 |
13 |
25 |
69.73 |
109.57 |
57.46 |
83.40 |
37.32 |
63.63 |
29.32 |
46.39 |
| LH (mIU/mL) |
1.76–10.24 |
1.5 |
9.1 |
1.5 |
9.0 |
0.07 |
1.03 |
0.03 |
0.3 |
0.02 |
0.35 |
0.04 |
0.24 |
| FSH (mIU/mL) |
3.21–14.72 |
3.5 |
12.0 |
5.4 |
16 |
0.44 |
3.14 |
0.26 |
2.08 |
0.66 |
2.97 |
0.30 |
1.72 |
| E2 (pg/mL) |
|
<10.0 |
N.A. |
N.A. |
N.A. |
<10.0 |
N.A. |
<10.0 |
N.A. |
<10.0 |
N.A. |
<10.0 |
N.A. |
| FT4 (ng/dL) |
0.70–1.48 |
0.93 |
N.A. |
N.A. |
N.A. |
0.67 |
N.A. |
0.97 |
N.A. |
0.67 |
N.A. |
0.86 |
N.A. |
| IGF-1 (ng/mL) |
147–358 |
N.A. |
N.A. |
N.A. |
N.A. |
29 |
N.A. |
57 |
N.A. |
64 |
N.A. |
118 |
N.A. |
| ITT |
Normal range |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
| ACTH (pg/mL) |
<46.0 |
26.0 |
81.0 |
28 |
257 |
12.9 |
16.6 |
<5.0 |
<5.0 |
<5.0 |
<5.0 |
<5.0 |
<5.0 |
| Cortisol (μg/dL) |
5.0–25.0 |
11.6 |
16.7 |
12.9 |
23.2 |
5.7 |
7.6 |
1.3 |
1.2 |
<1.0 |
<1.0 |
<1.0 |
<1.0 |
| GH (ng/mL) |
0.04–3.60 |
2.29 |
4.61 |
0.83 |
1.73 |
0.10 |
0.48 |
0.34 |
0.63 |
<0.05 |
0.15 |
0.63 |
0.85 |
| GHRP-2 loading test |
Normal range |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
Vor |
Peak |
| GH (ng/mL) |
0.04–3.60 |
N.A. |
N.A. |
N.A. |
N.A. |
0.43 |
0.63 |
0.21 |
1.85 |
0.05 |
0.63 |
0.54 |
0.99 |
| ACTH (pg/mL) |
<46.0 |
N.A. |
N.A. |
N.A. |
N.A. |
19.7 |
103.0 |
<5.0 |
12.6 |
<5.0 |
6.4 |
<5.0 |
14.2 |
| Cortisol (μg/dL) |
5.0–25.0 |
N.A. |
N.A. |
N.A. |
N.A. |
3.7 |
9.7 |
<1.0 |
1.3 |
<1.0 |
<1.0 |
<1.0 |
<1.0 |
Abbreviations: CAPST, combined anterior pituitary stimulation test (100 μg CRH, 100 μg GRH, 500 μg TRH, and 100 μg LHRH); ITT, insulin tolerance test; GHRP-2, growth hormone-related peptide-2; PSL, prednisolone; N.A., Not Available; p.o., per os

When she was 49 years old, she presented to our hospital with general fatigue and bitemporal hemianopia. T1-weighted MRI of the brain with heterogeneous gadolinium enhancement revealed swelling of the pituitary gland and stalk with a cystic appearance (Fig. 3). Pharmacologic loading tests showed an excessive response of ACTH to CRH (baseline ACTH 14.9 pg/mL, 30-min peak ACTH 203.0 pg/mL) and a low response to the ITT (baseline ACTH 12.9 pg/mL, peak ACTH 16.6 pg/mL). Laboratory findings also revealed secondary hypogonadism, secondary hypothyroidism and hyperprolactinemia. Furthermore, GH showed a low response to the ITT, fulfilling the criteria for AGHD (Table 2). Consequently, the patient was diagnosed with combined pituitary hormone deficiency.
We then compared her current state with a previous state when she was 35 years old, post-prednisolone treatment. Within 15 years, the cystic appearance in the sellar region had enlarged, resulting in pituitary stalk compression (Fig. 3). These changes led to an excessive response of PRL to TRH and a low response of ACTH and GH to the ITT (baseline ACTH 12.9 pg/mL, peak ACTH 16.6 pg/mL; baseline GH 0.10 ng/mL, peak GH 0.48 ng/mL) (Table 2). Since the responsiveness of ACTH to CRH remained (baseline ACTH 14.9 pg/mL, 30-min peak ACTH 203.0 pg/mL) (Table 2), we concluded that her hypopituitarism was mainly due to pituitary stalk compression.
To confirm the diagnosis, we performed a transsphenoidal pituitary biopsy. Pathological findings showed extensive fibrotic tissue, with few remnant ciliated columnar cells as well as nests of atrophic pituitary gland cells with scarce lymphocytic infiltration (Fig. 4). Based on these histopathological findings, she was diagnosed with hypophysitis secondary to RCC. The enlarged pituitary gland slightly shrank after biopsy, and the loading tests performed 2 months after biopsy with 20 mg/day hydrocortisone showed low baseline ACTH and cortisol levels, as well as a decrease in the ACTH and cortisol response to CRH (baseline ACTH <5.0 pg/mL, 120-min peak ACTH 49.7 pg/mL; baseline cortisol <1.0 pg/mL, 120-min peak cortisol 1.5 pg/mL) and the ITT (baseline ACTH <5.0 pg/mL, peak ACTH <5.0 pg/mL; baseline cortisol 1.3 pg/mL, peak cortisol 1.2 pg/mL). However, the ACTH response to GHRP-2 remained (baseline ACTH <5.0 pg/mL, peak ACTH 12.6 pg/mL) (Table 2).

She was treated with prednisolone (initial dosage 30 mg/day). The dosage of prednisolone was gradually reduced over the course of 3 months, with a total dose of 1,621 mg per os, and was changed to hydrocortisone (15 mg/day) (Fig. 3). Although the cyst volume further decreased rather than recovered after prednisolone treatment (Fig. 3), the ACTH, GH, TSH, LH, and FSH response was abolished according to the loading tests performed 1 month after prednisolone treatment (CRH stimulation test: baseline ACTH <5.0 pg/mL, 120-min peak ACTH 21.9 pg/mL, ITT: baseline ACTH <5.0 pg/mL, peak ACTH <5.0 pg/mL and GHRP-2: baseline ACTH <5.0 ng/mL, peak ACTH 6.4 ng/mL) (Table 2). The low response of ACTH and cortisol could be due to prednisolone loading since the evaluations were performed immediately after prednisolone treatment. To assess the endocrinological state of this patient without the effect of prednisolone, we re-evaluated the pharmacological tests 2.5 years after prednisolone treatment. ACTH responded to CRH (baseline ACTH <5.0 pg/mL, 120-min peak ACTH 55.7 pg/mL) and GHRP-2 (baseline ACTH <5.0 pg/mL, peak ACTH 14.2 pg/mL) but not to the ITT (baseline ACTH <5.0 pg/mL, peak ACTH <5.0 pg/mL) (Table 2). On the other hand, cortisol showed no response to CRH, the ITT, or GHRP-2 (Table 2).
Discussion
Rathke’s cleft cysts (RCCs) are known to be embryologic remnants of Rathke’s pouch and are non-neoplastic. RCCs are more frequently diagnosed currently, as imaging technology has developed and become widely available. Usually, RCCs are asymptomatic and seen in 13–22% of normal autopsies [15]. On the other hand, some RCCs are accompanied by various clinical features, such as headaches, visual disturbances, and hypopituitarism [16]. These clinical features are due to focal inflammation and mass effects. The long-term effectiveness of steroid treatment for symptomatic RCCs is still unknown.
Herein, we described the responses to prednisolone treatment in two cases of biopsy-proven secondary hypophysitis due to RCC. In both cases, prednisolone administration had an early effect of cyst shrinkage, followed by partial improvements in symptoms and pituitary functions. However, long-term observations showed that prednisolone treatment did not fully improve anterior pituitary hormone secretion.
For symptomatic RCCs, especially those with visual field defects, surgery is performed to drain the cyst content and to remove as much of the capsule as possible. After surgery, headaches and visual field defects improve in a significant number of patients (40–100% and 33–100%, respectively), and among the pituitary hormone abnormalities, hyperprolactinemia is most frequently resolved (in most series, 100%). However, among symptomatic RCC patients who undergo surgery, 4–30% show new anterior pituitary hormone deficits, and 2–67% of patients experience new onset of DI after surgery [17]. In the study by Aho et al., among patients who underwent complete removal of RCCs by trans-sphenoidal surgery, 98% showed improvements in visual disorders, 18% experienced recovery of gonadal function and GH secretion, and 14% showed improvements in ACTH function. On the other hand, 19% of the patients developed DI, and 18% of the patients showed recurrence within five years [7]. Taken together, these findings show that surgical intervention relieves symptoms, such as headaches and visual field disturbances, but has the possibility of causing postoperative endocrine dysfunction.
Recently, the administration of low-dose corticosteroids was reported to reduce the size of large RCCs [18]. Furthermore, relatively high-dose hydrocortisone was also reported to be effective in decreasing the size of RCCs. However, surgical removal of RCCs was eventually needed regarding the case in this report, since it was difficult to permanently continue high-dose glucocorticoid treatment [14]. Similarly, in Case 1, relatively high-dose hydrocortisone treatment reduced the size of RCC and demonstrated the recovery of pituitary function by alleviating the compression on the pituitary stalk due to RCC. In Case 2, the patient was treated with prednisolone at the former hospital under the diagnosis of lymphocytic hypophysitis, which also relieved the stalk compression due to RCC. These results suggest that glucocorticoids may be effective in some cases of RCC with suprasellar extension, especially in cases with stalk disturbances. However, re-enlargement of RCCs occurred in both cases after the discontinuation of glucocorticoids. By performing a pituitary biopsy, we were able to accurately diagnose the patients’ clinical states, ruling out possibilities such as infectious lesions and granulomatous diseases. The pathological results of the two cases not only revealed lymphocytic infiltration into the pituitary glands, which is a characteristic of lymphocytic hypophysitis, but also showed features of RCCs, such as ciliated columnar cells. Therefore, the patients in our cases were pathologically diagnosed with secondary lymphocytic hypophysitis due to RCCs.
Hypophysitis due to RCC is listed as one of the sella and parasellar diseases in the clinical guidelines of the Japan Endocrine Society [19]. Pathologically, hypophysitis is mainly classified into two forms: primary and secondary forms. The secondary form includes local lesions and systemic diseases [20]. Hypophysitis caused by RCC is defined as secondary hypophysitis due to the local lesion. Inflammation originating from RCC may cause granulomatous hypophysitis. Recently, Yang et al. reviewed eight patients with hypophysitis caused by ruptured RCCs [3-5, 14]. According to the review, the symptoms were DI (6 of 8 patients), visual disturbances (5 of 8 patients), and headaches (4 of 8 patients) [21]. Case 1 and Case 2 both had DI, which could be due to lymphocytic inflammation in the pituitary gland, and the visual disturbances seen in Case 2 could have been caused by the re-enlargement of RCC compressing the optic chiasm.
The mechanism of the size reduction of RCCs after glucocorticoid administration remains unclear. Maruyama et al., who presented the short-term effectiveness of glucocorticoids in reducing RCC size, proposed that glucocorticoids suppress the secretion of cyst fluid because of their anti-inflammatory effect on electrolyte regulation in epithelial cells [14]. However, he did not histologically suggest inflammation in the cyst wall. On the other hand, the RCC size can change in long-term observations as well. For instance, in a study over an average of 26.9 months, the size of RCCs decreased in 15.9% of cases, increased in 12.4%, and remained unchanged in 74.4% [22]. Since some of the cases of RCCs shrank naturally during the clinical time course, it may be difficult to clearly state the effect of relatively high amounts of corticosteroids on reducing the size of RCCs. However, because the natural reduction rate of RCCs is low and the size of RCCs decreases after short-term loading of steroids, glucocorticoids could be an effective treatment for symptomatic RCCs. In these two cases, since the enlarged pituitary glands shrank after biopsy and further diminished after prednisolone treatment, we would presume that both the decrease of the liquid contents and the improvement of the enlarged parenchyma contributed to the shrinkage of enlarged pituitary glands. Indeed, as shown in the MRI images of Case 1 and Case 2 the liquid content was removed, but not completely, after biopsy. In addition, it is noted that the swelling of the pituitary further recovered after prednisolone treatment (Fig. 1, Fig. 3). Taken together, we speculated that prednisolone is effective in both the shrinkage of cyst and the shrinkage of enlarged parenchyma.
Prednisolone treatment for secondary hypophysitis caused by RCCs is expected to exert an anti-inflammatory effect and to improve pituitary function, as seen in the treatment of lymphocytic hypophysitis [20]. It is difficult to improve the secretory function of all pituitary hormones, even for steroid-responsive lymphocytic hypophysitis [23]. Moreover, complete suppression of inflammation in RCCs is difficult even after prednisolone treatment, since the mucus secreted from the ciliated columnar cells of RCCs causes persistent pituitary inflammation. Therefore, the recovery of pituitary hormonal function may be more difficult in secondary hypophysitis due to RCCs than in lymphocytic hypophysitis. In Case 1, although prompt initiation of steroid therapy decreased the size of RCC, pituitary function did not recover completely after long-term follow-up. In Case 2, prednisolone was applied at the former hospital based on the diagnosis of lymphocytic hypophysitis, which partially improved ACTH and TSH secretion, but DI and hypogonadism remained. Even though prednisolone administration decreased the size of RCC, in the end, it did not result in full recovery of any of the pituitary hormones in either case. Even after optimal surgical resection, recovery of pituitary function is limited in most cases [17]. In fact, anterior pituitary function decreased in both of our cases even after pituitary biopsy. At the very least, in patients who cannot undergo surgery due to physical conditions, prednisolone is one of the options for reducing the size of RCCs and the partial recovery of pituitary function [14].
Last, we examined the possibility of autoimmunity as the pathogenesis of developing pituitary dysfunction in these two cases. It has been reported that pituitary antibodies are positive in some RCCs [24]. We measured anti-pituitary antibodies (APAs) in these two cases using indirect immunofluorescence (IIF). IIF is known as being low in sensitivity and low in quantity, and the analysis can be considered subjective; however, when the human pituitary gland is used as an IIF substrate, the sensitivity increases compared to other species, such as mice [24]. The APAs in these two cases were negative, but rat pituitary cells were used as the IIF substrate. Therefore, we cannot affirm that both of our cases were negative for APAs. Furthermore, we only have APA data from after glucocorticoid treatment in both of our cases; thus, the de novo state is unclear. Although we cannot completely deny the involvement of weak autoimmunity, based on the pathological and negative APA findings, we presume that the impaired anterior pituitary function is caused by secondary hypophysitis due to RCCs.
Since the etiology of hypophysitis due to RCCs has not been completely elucidated, further investigation is needed to confirm the effects of prednisolone treatment on hypophysitis due to RCCs.
Conclusion
In both of our cases of secondary hypophysitis caused by RCCs, prednisolone treatment had an early effect of cyst shrinkage, followed by a partial recovery of clinical symptoms and pituitary functions. Nevertheless, in the long term, prednisolone did not completely improve anterior pituitary hormone secretion.
Funding
This research was supported by Grants-in-Aid for Scientific Research (B) # 19H03708, (C) # 18K08464, # 18K08465, Fostering Joint International Research (A) #19KK0407, the Naito Foundation, the Takeda Science Foundation, GSK Japan Research Grant 2018 (higuchi), the Hamaguchi biochemistry Foundation, and the Yamaguchi endocrine Foundation. This work was supported by the Donated Fund of Next Generation Hormone Academy for Human Health & Longevity.
Conflict of Interest
The authors declare that they have no competing interests.
Research Involving Human Participants
All procedures were performed according to the Declaration of Helsinki, and this study was approved by the Committee on Ethics in Human Research of Chiba University. Written informed consent for publication of this case report was obtained from patients with hypophysitis caused by RCCs. All treatments were considered the standard of care.
Informed Consent
Written informed consent was obtained from the patients for publication of this case report, including any accompanying images. A copy of the written consent form is available for review by the Editor of this journal.
Authors’ Contributions
HD, HK, and TT designed and drafted the manuscript and interpreted the data. IS, SH, HN, NH, and KY participated in the endocrinological treatment and collected the data. GY, KH, and YI performed the transsphenoidal operation to obtain a biopsy from the pituitary gland. NI performed the pathological evaluations. All authors read and approved the final manuscript.
Abbreviations
ACTH, Adrenocorticotropin hormone; APA, Anti-pituitary antibody; CRH, Corticotropin releasing hormone; E2, Estradiol; FSH, Follicle-stimulating hormone; FT4, Free thyroxine; GH, Growth hormone; GnRH, Gonadotropin-releasing hormone; ITT, Insulin tolerance test; LH, Luteinizing hormone; PRL, Prolactin; RCC, Rathke’s cleft cyst; TRH, Thyrotropin-releasing hormone; TSH, Thyroid-stimulating hormone
References
- 1 el-Mahdy W, Powell M (1998) Transsphenoidal management of 28 symptomatic Rathke’s cleft cysts, with special reference to visual and hormonal recovery. Neurosurgery 42: 7–17.
- 2 Teramoto A, Hirakawa K, Sanno N, Osamura Y (1994) Incidental pituitary lesions in 1,000 unselected autopsy specimens. Radiology 193: 161–164.
- 3 Schittenhelm J, Beschorner R, Psaras T, Capper D, Nagele T, et al. (2008) Rathke’s cleft cyst rupture as potential initial event of a secondary perifocal lymphocytic hypophysitis: proposal of an unusual pathogenetic event and review of the literature. Neurosurg Rev 31: 157–163.
- 4 Nishikawa T, Takahashi JA, Shimatsu A, Hashimoto N (2007) Hypophysitis caused by Rathke’s cleft cyst. Case report. Neurol Med Chir (Tokyo) 47: 136–139.
- 5 Al Safatli D, Kalff R, Waschke A (2015) Spontaneous involution of a presumably Rathke’s cleft cyst in a patient with slight subclinical hypopituitarism: a case report and review of the literature. Case Rep Surg 2015: 971364.
- 6 Baskin DS, Wilson CB (1984) Transsphenoidal treatment of non-neoplastic intrasellar cysts. A report of 38 cases. J Neurosurg 60: 8–13.
- 7 Aho CJ, Liu C, Zelman V, Couldwell WT, Weiss MH (2005) Surgical outcomes in 118 patients with Rathke cleft cysts. J Neurosurg 102: 189–193.
- 8 Billeci D, Marton E, Tripodi M, Orvieto E, Longatti P (2004) Symptomatic Rathke’s cleft cysts: a radiological, surgical and pathological review. Pituitary 7: 131–137.
- 9 Han SJ, Rolston JD, Jahangiri A, Aghi MK (2014) Rathke’s cleft cysts: review of natural history and surgical outcomes. J Neurooncol 117: 197–203.
- 10 Mendelson ZS, Husain Q, Elmoursi S, Svider PF, Eloy JA, et al. (2014) Rathke’s cleft cyst recurrence after transsphenoidal surgery: a meta-analysis of 1151 cases. J Clin Neurosci 21: 378–385.
- 11 Ogawa Y, Watanabe M, Tominaga T (2011) Prognostic factors of operated Rathke’s cleft cysts with special reference to re-accumulation and recommended surgical strategy. Acta Neurochir (Wien) 153: 2427–2433.
- 12 Ogawa Y, Watanabe M, Tominaga T (2013) Rathke’s cleft cysts with significant squamous metaplasia—high risk of postoperative deterioration and close origins to craniopharyngioma. Acta Neurochir (Wien) 155: 1069–1075.
- 13 Potts MB, Jahangiri A, Lamborn KR, Blevins LS, Kunwar S, et al. (2011) Suprasellar Rathke cleft cysts: clinical presentation and treatment outcomes. Neurosurgery 69: 1058–1068.
- 14 Maruyama H, Iwasaki Y, Tsugita M, Ogami N, Asaba K, et al. (2008) Rathke’s cleft cyst with short-term size changes in response to glucocorticoid replacement. Endocr J 55: 425–428.
- 15 Raper DM, Besser M (2009) Clinical features, management and recurrence of symptomatic Rathke’s cleft cyst. J Clin Neurosci 16: 385–389.
- 16 Komatsu F, Tsugu H, Komatsu M, Sakamoto S, Oshiro S, et al. (2010) Clinicopathological characteristics in patients presenting with acute onset of symptoms caused by Rathke’s cleft cysts. Acta Neurochir (Wien) 152: 1673–1678.
- 17 Trifanescu R, Ansorge O, Wass JA, Grossman AB, Karavitaki N (2012) Rathke’s cleft cysts. Clin Endocrinol (Oxf) 76: 151–160.
- 18 Furtado SV, Venkatesh PK, Ghosal N, Hedge AS (2010) Reduction in size of a large Rathke’s cleft cyst on treatment with low dose of corticosteroid. Horm Metab Res 42: 227–229.
- 19 Takagi H, Iwama S, Sugimura Y, Takhashi Y, Oki Y, et al. (2020) Diagnosis and treatment of autoimmune and IgG4-related hypophysitis: clinical guidelines of the Japan Endocrine Society. Endocr J 67: 373–378.
- 20 Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, et al. (2005) Autoimmune hypophysitis. Endocr Rev 26: 599–614.
- 21 Yang C, Wu H, Bao X, Wang R (2018) Lymphocytic hypophysitis secondary to ruptured Rathke cleft cyst: case report and literature review. World Neurosurg 114: 172–177.
- 22 Sanno N, Oyama K, Tahara S, Teramoto A, Kato Y (2003) A survey of pituitary incidentaloma in Japan. Eur J Endocrinol 149: 123–127.
- 23 Koide H, Shiga A, Komai E, Yamato A, Fujimoto M, et al. (2018) Prednisolone-responsive postpartum IgG4-related hypophysitis. Intern Med 57: 367–375.
- 24 Ricciuti A, De Remigis A, Landek-Salgado MA, De Vincentiis L, Guaraldi F, et al. (2014) Detection of pituitary antibodies by immunofluorescence: approach and results in patients with pituitary diseases. J Clin Endocrinol Metab May 99: 1758–1766.
