特集号: 鉄と鋼
107 巻, 12 号
特集号「インフラ構造物の経年劣化に対する維持管理の最適化に向けて」
選択された号の論文の15件中1~15を表示しています
- |<
- <
- 1
- >
- >|
出版情報
-
2021 年 107 巻 12 号 p. Cover-
発行日: 2021/12/01
公開日: 2021/11/30
PDF形式でダウンロード (668K) -
2021 年 107 巻 12 号 p. Contents-
発行日: 2021/12/01
公開日: 2021/11/30
PDF形式でダウンロード (3031K) -
2021 年 107 巻 12 号 p. Editorial-
発行日: 2021/12/01
公開日: 2021/11/30
PDF形式でダウンロード (205K)
特集号「インフラ構造物の経年劣化に対する維持管理の最適化に向けて」
-
2021 年 107 巻 12 号 p. 997
発行日: 2021年
公開日: 2021/11/30
PDF形式でダウンロード (258K) HTML形式で全画面表示 -
2021 年 107 巻 12 号 p. 998-1003
発行日: 2021年
公開日: 2021/11/30
 PDF形式でダウンロード (1446K) HTML形式で全画面表示
PDF形式でダウンロード (1446K) HTML形式で全画面表示 -
2021 年 107 巻 12 号 p. 1004-1010
発行日: 2021年
公開日: 2021/11/30
 PDF形式でダウンロード (1436K) HTML形式で全画面表示
PDF形式でダウンロード (1436K) HTML形式で全画面表示 -
2021 年 107 巻 12 号 p. 1011-1019
発行日: 2021年
公開日: 2021/11/30
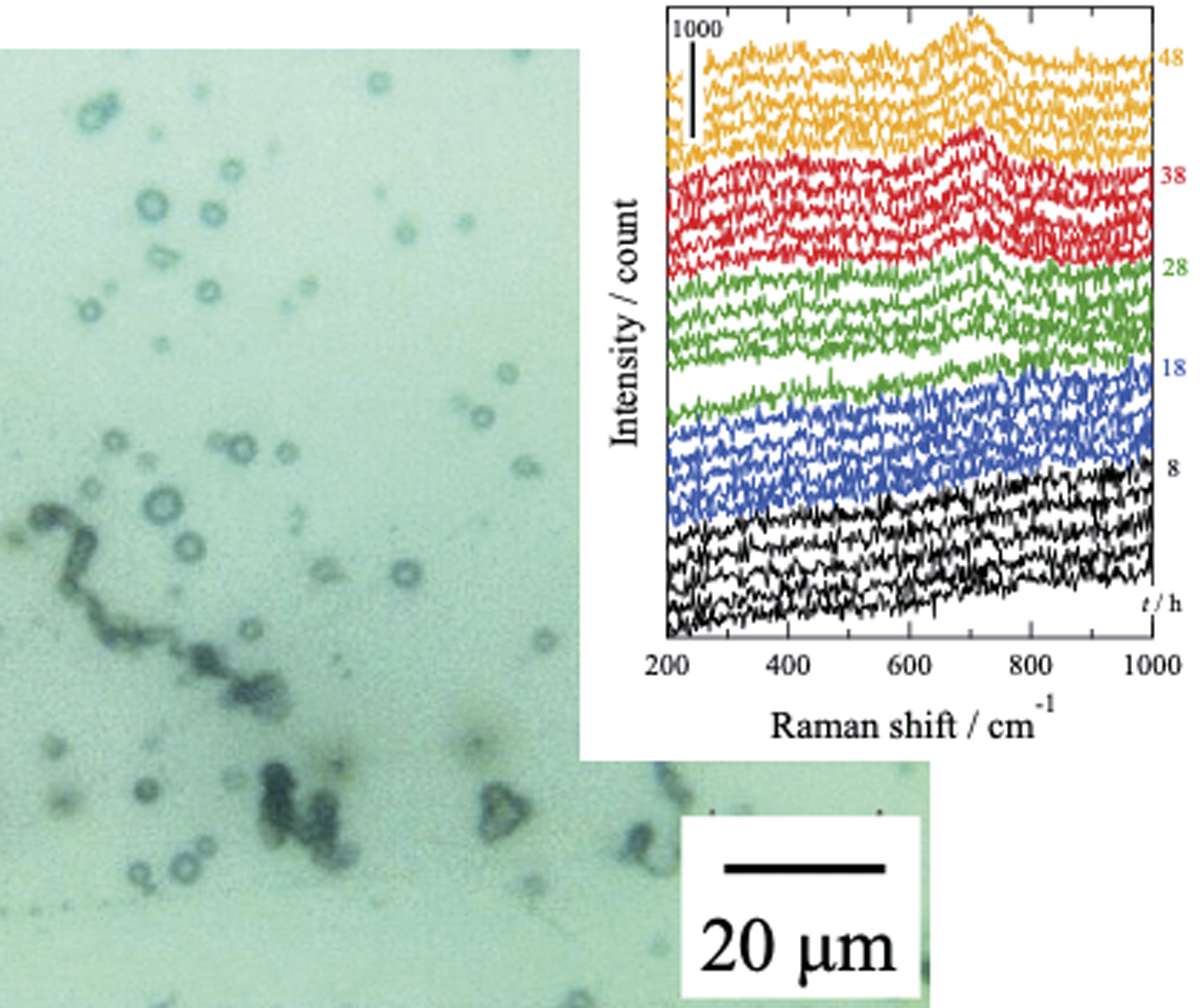 PDF形式でダウンロード (2490K) HTML形式で全画面表示
PDF形式でダウンロード (2490K) HTML形式で全画面表示 -
2021 年 107 巻 12 号 p. 1020-1028
発行日: 2021年
公開日: 2021/11/30
[早期公開] 公開日: 2021/03/19 PDF形式でダウンロード (16194K) HTML形式で全画面表示
PDF形式でダウンロード (16194K) HTML形式で全画面表示 -
2021 年 107 巻 12 号 p. 1029-1035
発行日: 2021年
公開日: 2021/11/30
 PDF形式でダウンロード (1237K) HTML形式で全画面表示
PDF形式でダウンロード (1237K) HTML形式で全画面表示 -
2021 年 107 巻 12 号 p. 1036-1046
発行日: 2021年
公開日: 2021/11/30
[早期公開] 公開日: 2021/06/08 PDF形式でダウンロード (5509K) HTML形式で全画面表示
PDF形式でダウンロード (5509K) HTML形式で全画面表示 -
2021 年 107 巻 12 号 p. 1047-1056
発行日: 2021年
公開日: 2021/11/30
 PDF形式でダウンロード (6707K) HTML形式で全画面表示
PDF形式でダウンロード (6707K) HTML形式で全画面表示 -
2021 年 107 巻 12 号 p. 1057-1065
発行日: 2021年
公開日: 2021/11/30
 PDF形式でダウンロード (12231K) HTML形式で全画面表示
PDF形式でダウンロード (12231K) HTML形式で全画面表示 -
2021 年 107 巻 12 号 p. 1066-1073
発行日: 2021年
公開日: 2021/11/30
 PDF形式でダウンロード (5060K) HTML形式で全画面表示
PDF形式でダウンロード (5060K) HTML形式で全画面表示 -
2021 年 107 巻 12 号 p. 1074-1084
発行日: 2021年
公開日: 2021/11/30
 PDF形式でダウンロード (8024K) HTML形式で全画面表示
PDF形式でダウンロード (8024K) HTML形式で全画面表示 -
2021 年 107 巻 12 号 p. 1085-1094
発行日: 2021年
公開日: 2021/11/30
[早期公開] 公開日: 2021/05/15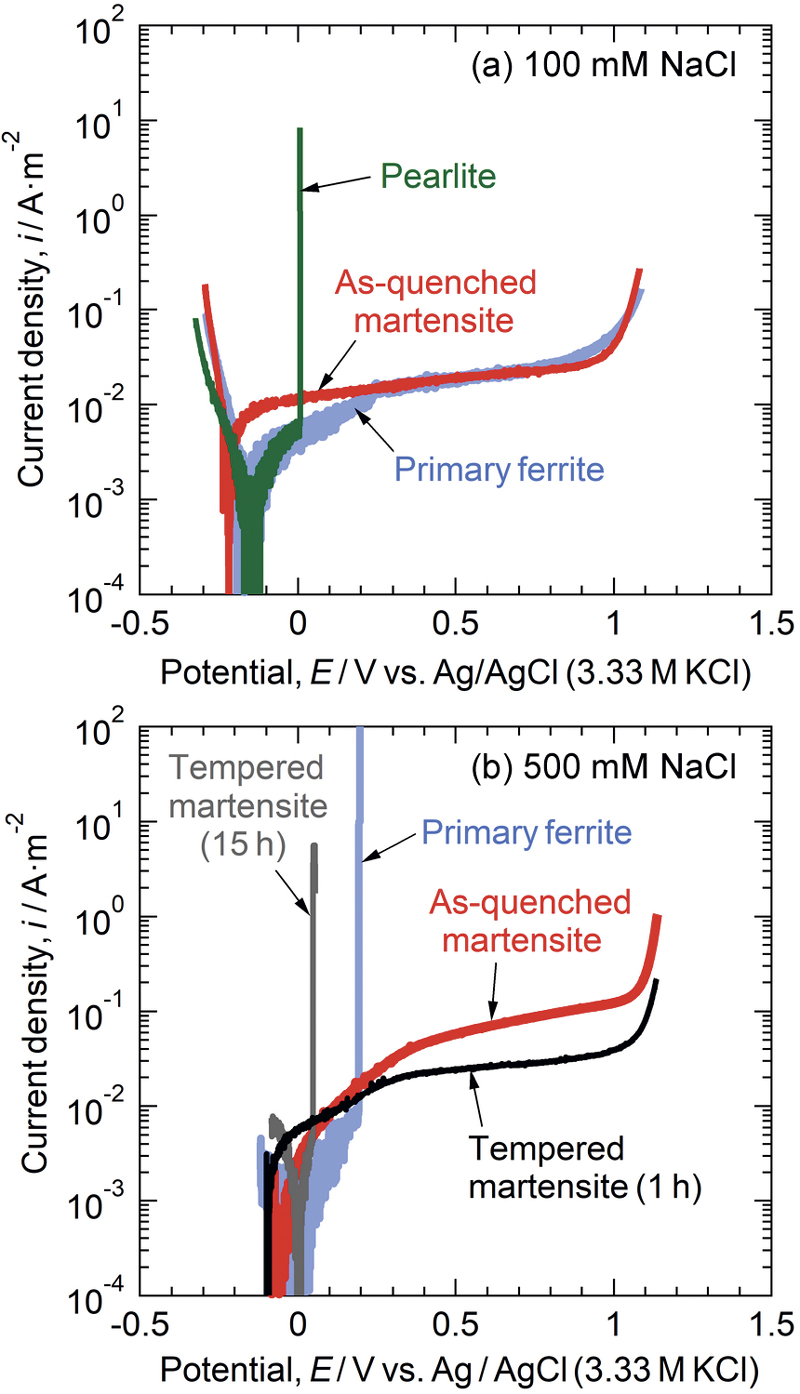 PDF形式でダウンロード (7187K) HTML形式で全画面表示
PDF形式でダウンロード (7187K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|