速報
COVID-19の経口治療薬開発に向けたハイブリッド型in Silico創薬
2022 年 21 巻 2 号 p. 48-51
詳細
2022 年 21 巻 2 号 p. 48-51
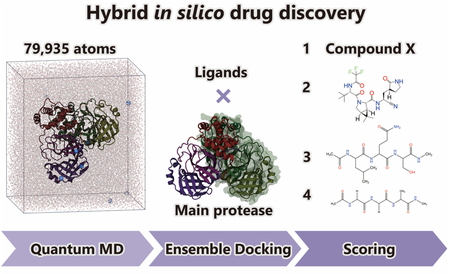
Hybrid in silico drug discovery was performed by combining large-scale quantum molecular dynamics (QMD) simulations with the conventional in silico drug discovery, focusing on developing covalent inhibitors against the main protease (Mpro) of SARS-CoV-2, the virus responsible for ongoing COVID-19 pandemic. The crystal structures and instantaneous structures obtained from the large-scale QMD simulations for Mpro were used as receptors in ensemble docking to estimate the binding affinities of the four ligands: the natural substrate recognized by Mpro, that recognized by the other enzyme of SARS-CoV-2, approved covalent inhibitor (PF-07321332), and the new candidate compound X determined from virtual screening. The present result shows that the binding affinity of X was comparable to that of PF-07321332, demonstrating the potency of our drug discovery.