Review Papers
Deficiencies in Traditional Oral Dosage Forms and the Emergence of Controlled-Release Powder Manufacturing
2017 Volume 34 Pages 91-105

Details
2017 Volume 34 Pages 91-105
The importance of providing safe and effective delayed- and extended-release oral formulations that can replace products requiring multiple administrations has been continually cited as an area in need of improvement for pharmaceutical companies. Such controlled release challenges become especially critical when they must be adapted for paediatrics, those suffering from dysphagia, or patients with specific dosage administration limitations. More often than not, lack of palatability and taste-masking compound this formulation challenge. Many particulate approaches show promise, but can be fraught with broad particle size distributions, initial drug burst, poor drug entrapment efficiency, low drug loading, and limited scalability. Here, we summarize the key factors that drive formulation development of format-flexible controlled-release oral powders, and the manufacturing aspects involved with some of the foremost marketed products, including next-generation single-step layered powder manufacturing (below).
The importance of providing safe, effective, and proven medicines for populations with dysphagia, such as children, geriatrics, and those suffering from debilitating illnesses, has been continually cited as an area in need of improvement for pharmaceutical companies and the providers who administer their product (Bergstrom D. et al., 2004; Bhardwaj S. and Hayward M., 1996; Cram A. et al., 2013; Dickens D.S. et al., 2008; Engelen L. et al., 2005; Imai E. et al., 1995; Ivanovska V. et al., 2014; Lopez F.L. et al., 2015; Matsui D., 2007; Milne C.P. and Bruss J.B., 2008; Rocca J.G. and Park K., 2004; Sugao H., 1997; Tyle P., 1993). The widespread lack of dispersed format oral products, however, forces clinicians and pharmacists to use alternative solutions to treat their patients that are not always backed by supporting bioavailability, stability, and safety studies. Tablets are sometimes administered extemporaneously by crushing the dosage form and mixing with food or drink. Not only are these delivery methods inconsistent, they often lead to dosing errors, decreased bioavailability or efficacy, and non-adherence because of foul-tasting active pharmaceutical ingredients (APIs) (Jayanthi B. and Manna P., 2011; Osterberg L. and Blaschke T., 2005; Sansom L., 1999; Schier J. et al., 2003).
Due to taste and efficacy concerns, The Institute for Safe Medical Practices (ISMP) has issued a “Do Not Crush” list, which highlights over 400 dosage forms that cannot be compounded due to special controlled-release properties, taste-masking, or API protection (Bergstrom D. et al., 2004; Bhardwaj S. and Hayward M., 1996; Dickens D.S. et al., 2008; Engelen L. et al., 2005; Imai E. et al., 1995; Matsui D., 2007; Milne C.P. and Bruss J.B., 2008; Rocca J.G. and Park K., 2004; Sugao H., 1997; Tyle P., 1993). Lack of titratable and palatable formulations affects over half of the global population (under 18 and over 65 years of age) and can subject patients to avoidable adverse events (Jayanthi B. and Manna P., 2011; Osterberg L. and Blaschke T., 2005; Sansom L., 1999; Schier J. et al., 2003). Of all medication-related hospitalizations that occur in the United States, between one-third and two-thirds are the result of poor medication adherence (Matsui D., 2007; Milne C.P. and Bruss J.B., 2008). Current dosing regimens for populations with dysphagia or those unwilling to take traditional tablets or capsules suffer because many current oral formulations fail to simultaneously address the critical aspects discussed below (Griffith R., 2005; Wright D., 2002).
1.1 PalatabilityMany APIs are extremely bitter and coating efforts can be ineffective or result in an unpleasant mouth feel due to irregular surface features. Approximately half of patients with organoleptic sensitivities refuse to take their medicine, with the large majority of those reporting bad taste as the single major reason for non-compliance (Matsui D., 2007). Sweeteners alone are often unable to overcome the extremely unpleasant taste of many active ingredients in syrups and suspensions (Bergstrom D. et al., 2004; Bhardwaj S. and Hayward M., 1996; Dickens D.S. et al., 2008). Moreover, efforts to mask flavors using coatings, agglomeration, or microencapsulation often result in poorly-controlled, heterogeneous particle distributions that result in a gritty or granular mouth feel. Ideally, a highly palatable dosage form will consist of taste-masking with a smooth mouthfeel while maintaining other controlled release properties.
1.2 Titratable dosingDifferent ages, weights, body mass indices, and metabolically-impaired individuals require considerable dosing precision that is not linearly scaled (Milne C.P. and Bruss J.B., 2008). Because medication errors are common in six percent of pediatric hospitalizations, dose titration is critical, as a “one-size-fits-all” dosing is ineffective in children due to their developmental variability (Cram A. et al., 2013; Ivanovska V. et al., 2014; Lopez F.L. et al., 2015). and can prove deleterious for geriatric patients with hepatic or renal impairment.
1.3 Controlled-release kineticsAchieving controlled-release kinetics with tablets is a relatively simple process, as the size and form factor of the dosage form lends to using robust coating methods, sometimes with several layers (Jayanthi B. and Manna P., 2011; Sansom L., 1999). Capsules have the advantage of being injection moulded, extruded, or die pressed with gelatines and other controlled-release polymers in a repeatable, high-throughput manner, enabling large doses of medication in a modest form factor. Tablets are simply pressed by traditional means, then coated with subsequent layers of controlled-release components, which makes translation of specialised functions (e.g. delayed or extended release) simple with a pill format (Jayanthi B. and Manna P., 2011; Sansom L., 1999). The major problem, however, is the size of such dosage forms, which renders them impracticable to certain patient populations.
Universal technology hurdles exist independently of drug or indication even for adult populations, and thus substantially impede development of optimal therapeutics for those with special dosing considerations. Specifically, for pediatric drugs, companies must meet all standard pharmaceutical benchmarks (bioavailability, shelf life, safety, and efficacy) but also consider palatability, dosing accuracy, and age-appropriate formats. Through the Best Pharmaceuticals for Children Act, Pediatric Research Equity Act, and EMEA Pediatric Investigation Plans, the industry is either incentivized or in some cases required to perform pediatric studies (2002, 2003, 2006; Cram A. et al., 2013). Internal capabilities for this development are limited, and many companies do not have the capacity to make pediatric formulations. This emphasizes the need for scalable, flexible, cost-effective formulation platforms for practical, palatable, age-appropriate, and accurate dosing to create safer medications that enhance compliance at any age. While advances in pediatric-centric dosage formats take to market, adults with dysphagia will also secure the benefits of research and development efforts focused on providing alternative formats to historical solid oral dosage forms.
2.1 Size burden of tablets and capsulesData from marketed pharmaceutical products indicates that the average size of a controlled- or extended-release tablet is nearly 1.5 cm in length (Pharmacircle, 2016). Physiological studies demonstrate that swallowing becomes increasingly difficult when the dimension of the object being ingested reaches more than 50 % of the oesophageal diameter, which is 2.0 cm for the prototypical adult.(Harb J., 2015) By this logic, the average controlled-release tablet is too large to be swallowed comfortably. The merits residing in pills and capsules is that they contain the volumetric space to (1) deliver a large payload of API, (2) utilize elaborate controlled-release mechanisms (such as ion-exchange, micro-pumping mechanisms, etc.), and (3) circumvent many shelf stability challenges. These merits, however, fall short of benefitting a significant fraction of the world population simply due to the form factor size.
2.2 Taste and consistency burden of syrups, suspensions, elixirs, and solutionsWhere large tablets and capsules present swallowing and administration challenges, liquid formats succeed in dose titration most of the time. The advantages beyond ease of dosing are limited in traditional syrups, however. Liquid formats (1) are not extended-release, (2) have little- to-no taste-masking, (3) and can contain API particles prone to settling and aggregation if not reconstituted properly prior to administration, which have resulted in risks to patient safety (Bergstrom D. et al., 2004; Cram A. et al., 2013; Ivanovska V. et al., 2014; Lopez F.L. et al., 2015; Matsui D., 2007; Milne C.P. and Bruss J.B., 2008). Recent advances (which will be discussed later in this article) have enabled extended release and taste-masking of orally-administered APIs, but the breadth of application currently covers less than one percent of marketed drugs.
2.3 Alternatives to traditional dosage formsAs a technical resolution to the large format of controlled-release pills and capsules, the foul taste of traditional syrups and suspensions, and the lack of controlled-release options for APIs tableted and encapsulated in nearly 85 % of marketed drugs (Table 1) (Maalouf N., 2013; Vummaneni V. and Nagpal D., 2012), many pharmaceutical and contract manufacturing organizations (CMOs) are focusing research and development efforts on controlled-release powder formats, which combine the stability of solid oral dosage forms and dose titration advantages of liquids. These tablet alternatives address many of the deficiencies discussed earlier, but can still be fraught with inadequacies such as multiple-step manufacturing and inconsistent particle sizes.
| Types of Pediatric/Geriatric/Dysphagia-Friendly Formulations | Of Most-Prescribed Drugs (%) | Taste Masking Methods | Disadvantage | Advantage | Major Technologies and Companies Practicing in Each Dosage form Space |
|---|---|---|---|---|---|
| Suspension | 13.0 | coating, microencapsulation, ion exchange resin, inclusion complexes, granulation, adsorption, prodrug, bitterness inhibitors, multiple emulsion, gel formation | Easy to swallow, can be taste-masked, though most form factors of these typs are not (see exceptions at right). | TPG/Adare: Microcaps®, Liquitard®. Tris: OralXT™, LiquiXR™ Cipla: Sprinkles®, Orbis: Optimμm® | |
| Granules/Powder | 2.8 | ||||
| Orally Disintegrating Tablet (ODT) | 3.0 | TPG/Adare: AdvaTab®, Diffucaps®. Tris: ODTXR™. SPI: Actimask®. Catalent: Zydis®. Cima: OraSolv®, Durasolv®, Lyoc™ | |||
| Chewable Tablet | 4.2 | Tris: ChewableXR™. SPI: Actimask® | |||
| Sublingual/Lingual | 0.6 | Tris: StripsXR™ | |||
| Tablet | 42.7 | Does not help those with dysphagia | Taste-masking available | TPG/Adare, Catalent, Capsugel/Bend Reserach, Allergan/Actavis, Pfizer, GlaxcoSmithKline, Bayer, Procter & Gamble, Johnson &Johnson, Bristol Myers Squibb, AstraZeneca, Impax, SPI, | |
| Capsule | 18.0 | ||||
| Solution/Syrup | 15.0 | Inherently no taste-masking: drug freely/rapidly available | No swallowing difficulty for those with dysphagia | ||
| Elixir | 0.8 | ||||
Micro- and nanoparticulate powders are manufactured with a myriad of processes, but the primary motivation is integration of controlled-release mechanisms to govern particle disintegration and API dissolution. The requirement of achieving controlled release universally relies on physical sequestration of the API via one or more physicochemical mechanisms, which typically requires multiple steps. A powder form factor, however, can present unique challenges to achieving controlled release coatings due to (1) the high surface area of particles, (2) irregular sizes of particles within the powder, and (3) the number of process steps required to ensure predictable performance and reasonable quality of the final product. Though, taste-masking can still be achieved with powders when a coating or other chemical modification is applied. Here, we discuss the salient manufacturing steps for current and next-generation dosage forms (Table 2) (Cram A. et al., 2013; Maalouf N., 2013; Vummaneni V. and Nagpal D., 2012).
| Powder-Based Technology | Manufacuring Considerations | Form Factor | Proprietor of Technology | Flagship Formulations Approved or In Pipeline |
|---|---|---|---|---|
| Optimμm® | Single-step microencapsulation, uniform particle size distribution (100s–1000s μm), scalable, dosage and format flexible, enables combination therapy | Microspheres and Microcapsules | Orbis Biosciences | predninsone, guaifenesin, ibuprofen |
| DETERx® | Multi-step production (drug-wax complex, particle fabrication, coating), dosage and format flexible, less uniform size distribution, abuse deterrent formulation | Hydrophobic matrix as physical or diffusional barrier | Collegium Pharmaceutical | oxycodone, oxymorphone, hyrocodone, morphine, methylphenidate |
| LiquiTime® | Multi-step production (granulation, coating, drying), non-uniform granule size distribution, size limited (> 100 um), dosage and format flexible | Traditional 2-Step Coating of particles | Flamel Technologies | guaifenesin, ibuprofen |
| Microcaps® (Used alone and in Liquitard®, AdvaTab® | API particles complexed with comtrolled-release matrix | Drug-coated powder | Adare Pharmaceuticals | tenofovir disoproxil fumarate, potassium chloride, paracetamol |
| Diffucaps® | Multi-step production (granulation, coating, drying), non-uniform granule size distribution, size limited (> 500 μm), dosage and format flexible | Minitablets | Adare Pharmaceuticals | propranalol, methylphenidate, cyclobenzaprine |
| Sprinkles® | Minitablets | Cipla | lopinavir, ritonavir | |
| OralXR+™ (Used alone and in LiquiXR™, ChewableXR™, ODTXR™, StripsXR™) | Multi-step production (resin priming, drug loading, resinate wash, drying), requires acidic or basic drug chemical structure, relies on proprietary resin beads | Solid suspension or ODT | Tris Pharma | amphetamine, codeine/chlorpheniramine polistirex, dextromethorphan |
| XR Technologies (RDIM, DTRS®, and KCTP™) | Neos Therapeutics | methylphenidate | ||
| ORaSolv®, DuraSolv®, Lyoc™ | API granules co-lyophilized with matrix | ODT | Cima Labs | loperamide, clozapine, prednisolone |
| Actimask® | ODT | SPI Pharmaceuticals | paracetamol, ibuprofen | |
| Zydis® | ODT | Catalent Pharma Solutions | lorazepam, loratadine, piroxicam, olanzapine |
The most straightforward method for achieving taste-masking and controlled release with powders employs a two-step process in which a precursor particle is manufactured by various means, then coated with one or more layers containing controlled-release materials (Fig. 1).

Powder creation and coating has traditionally been at least a two-step process, but provides great flexibility in end product dosage form and function. Each technique gives a different particle size range, with vibratory techniques creating the largest powders, and spray drying the smallest powders.
Precursor particles can either be (1) finely-milled API crystals, (2) crystalline or amorphous API co-mixed with inert bases or controlled-release materials, or (3) an entirely inert core without API. These three types of precursors can be manufactured by one or more means, which include traditional vibratory methods, congealing/spinning disk atomization, prilling, hot-melt extrusion (HME) and spheronization, aqueous dispersion, blending/bulking, electrohydrodynamic spraying (EHDS), or spray drying (Ambike A.A. et al., 2005; Cloupeau M. and Prunet-Foch B., 1994; Eldem T. et al., 1991; Gharsallaoui A. et al., 2007; Hancock B.C. et al., 2003; Passerini N. et al., 2006; Vehring R., 2007; Yurteri C.U. et al., 2010). Material selection for the precursor particle relies on process capabilities, desired end-product controlled-release properties, API thermal and oxidative stability and desired physical properties (surface features, density, friability, hardness, etc). Once the precursor microparticulate is manufactured, the product, in some instances, can be considered “finished.”
If taste-masking, delayed-release, or stability-enabling properties are required, the precursor particle advances to subsequent traditional coating steps using fluidized beds, Würster coaters, dry polymer coating, spray coating, pan coating, or coacervation (Dewettinck K. and Huyghebaert A., 1999; Gouin S., 2004; Jono K. et al., 2000; Lopez F.L. et al., 2015; Sastry S.V. et al., 2000). Materials of choice for the secondary coating steps are selected for reasons commensurate with precursor particles (i.e. material compatibility, controlled-release behaviour, and stability). The final dosage form can then be bottled for resuspension at time of use, packaged in sachets or sprinkle capsules, placed in dissolving tongue strips, co-lyophilized with other materials for orally-disintegrating tablets (ODTs), or reconstituted in syrup if liquid stability is not an issue (Table 2).
The history of manufacturing controlled-release powders by adding one or more coating steps to API-rich cores is no less than colossal, and for this reason we have provided a high-level overview with references to detailed reviews. These techniques are, however, divergent from state-of-the art techniques that focus on chemical modification of the API and/or substrate to which it is affixed via ion exchange resins (Elder D.P., 2005; Fazal U.-R. and Khan S.N., 2012; Pande S.V. et al., 2011). The main advantages that these methods can yield are (1) enabling liquid stability, and (2) deterring abuse of scheduled APIs, such as opiates and amphetamines. While revolutionary, drug complexation employs a number of manufacturing steps that far surpasses that of simple bead layering, and include and still usually include a final coating step (Dewettinck K. and Huyghebaert A., 1999; Gouin S., 2004; Jono K. et al., 2000; Lopez F.L. et al., 2015; Sastry S.V. et al., 2000). Here, we look in detail at two processes.
3.2 Granules with increased lipophilicityCollegium Pharmaceutical’s manufacturing process achieves extended release with abuse-deterrent properties via two major steps (1) API solubility reduction and (2) API carrier dispersion. (Eldem T. et al., 1991; Passerini N. et al., 2006) An optional third step is, unequivocally, coating for added protection of the particle contents.
Solubility reduction can be achieved by either reducing the overall charge of the API molecule, forming a salt between the API and a lipid moiety, forming a complex between and API and metal cation, complexation with cyclodextrins, or covalently modifying the molecule with ester or amide linkages.
Following solubility reduction, the modified API is dissolved in a molten wax or lipid with or without the assistance of co-solvents. Suitable materials include fatty alcohols, fatty esters, fatty acid glycerides, hydrogenated oils, and stearates. The final powder is then made via traditional means using congealing, extrusion/spheronization, prilling, or aqueous dispersion (Fig. 1) (Eldem T. et al., 1991; Passerini N. et al., 2006). Optional coatings can then be added by traditional means used for coated drug granules with one or more controlled-release polymers for taste-masking or API protection (Dewettinck K. and Huyghebaert A., 1999; Gouin S., 2004; Jono K. et al., 2000; Lopez F.L. et al., 2015; Sastry S.V. et al., 2000).
3.3 Ion exchange resin complexationSimilar to Collegium’s DETERx technology, Tris Pharma and Neos Therapeutic’s technologies also use a drug-tethering concept, but also provide liquid stability and extended-release. The manufacturing methods between Tris and Neos are very similar (Fig. 2), but are described here in brief. The main premise with these two technologies is reliance on an ion-exchange complex between API and inert substrate (Elder D.P., 2005; Fazal U.-R. and Khan S.N., 2012; Mehta K. et al., 2012; Pande S.V. et al., 2011).

Tris, Neos, and Collegium all use ion-exchange type processes for powder precursors, but Collegium disperses the API-complex in lipids and waxes that constitute the particle bulk, whereas Tris and Neos place thin coatings over the API-resin complex. All three techniques provide controlled-release, taste-masking, and abuse deterrence.
The first major step includes manufacturing of a resin microparticulate with charged moieties. Resin particles can be created through traditional processes such as suspension polymerisation with polystyrene, methacrylate, or acrylonitrile salts or variants thereof. Resins can be cationic or anionic in nature, depending on whether the API to be complexed is weakly or strongly acidic or basic. The resin must then be primed to ion exchange with the API.
The second step incorporates the ion exchange process during which the API of interest is ionized by salt removal, then complexed to the resin microparticle precursor by itself or with other additives that enhance stability or solubility, such as sugars. This process creates a bond between the API and resin that is more physically and chemically robust than mixing and emulsifying processes in traditional powder extrusion and layering discussed previously. Following this, the resinate is (1) washed to eliminate unreacted API, then (2) dried for the final formulary step.
Just as in traditional powder-coating methods, a final step focuses on securing the complexed API to the resin by coating with controlled-release polymers that provide diffusional control or pH-responsiveness, or layered with lipids and waxes. Following the drug-resin coating, the powder is ready for final format packaging, which may include compressing in to ODTs, filling in sachets, titrating into capsules, or bottling into liquid suspensions. Because taste is only perceived when the API is in solution, ion exchange resins provide taste-masking because the API is sequestered to the inert substrate.
3.4 Next generation powder manufacturingDeveloping controlled-release powders, to date, has employed combinations of manufacturing mechanisms and complex chemistry, which achieve substantial advantages over traditional pill and capsule formats, enabling extended- and delayed-release liquid suspensions and powder sachets, while providing taste-masking as-is, in a liquid constituent, or further compounded into ODTs or dissolving strips. The major criticisms of these methods, however, focus on the sheer number of process steps and excipients with undefined long-term safety and toxicology profiles (such as polystyrene). Thus, it comes as no surprise that manufacturers are investigating less complex chemistry and single-step manufacturing methods for producing controlled-release powders.
One such technology platform, Precision Particle Fabrication technology, also known as Optimμm technology, comes from Orbis Biosciences. The manufacturing scheme creates microsphere and microcapsule powders with high drug loading while maintaining the ability to process into a variety of extended-release, delayed-release, and taste-masked formats (Berkland C. et al., 2007a, 2007b; Pack D.W. et al., 2004a, 2003, 2001, 2002, 2004b).
The approach first creates either (1) a homogenous API-excipient mixture or suspension (i.e. for non-layered microspheres), or (2) separate core and drug-free shell solutions (i.e. for coated microspheres) (Fig. 3). High-shear mixing under controlled heat is added, after which the contents are transferred through heat-traced process lines and sprayed through a nozzle with (1) vibrational excitation to produce uniform droplets and (2) stabilizing anti-solvent stream to reduce the diameter of the exiting jet. The platform is capable of processing a variety of liquids, melts, polymer mixtures, slurries, and solutions of various viscosities and thermal properties. Unlike other encapsulation techniques, Optimμm’s central advantage is the ability to, in a single step, manufacture multi-kilogram batches of microcapsules (core and coating) having nominal diameters around 100 μm and API content up to 80 % by volume. Controlled-release formulations in the pipeline using this next-generation powder technology are APIs such as Ibuprofen, Guaifenesin, Prednisone, and Ritonavir, where the titratable dosing and flexibility allows for unparalleled dissolution matching against reference listed drugs (RLDs). The simultaneous advantages of this manufacturing method are (1) production of uniform, monodisperse powders, (2) single-step manufacturing, (3) the ability to control a wide range of therapeutic classes, (4) no requirement for API modification or complexation, (5) no requirement for processing solvents, (6) controlled-release with high API content, and (7) particle sizes that are indiscernible to the patient.

Precision Particle Fabrication Technology, also known as Optimμm technology, creates monodisperse powders just as traditional vibratory methods, but with (1) highly reduced diameters and (2) single-step manufacturing of coatings, which conserves time and resources of historical powder manufacturing techniques. There is also no need for complex chemistry beyond the controlled-release polymers used. Depending on the formulation steps used to incorporate the API, abuse deterrence with Optimμm technology is possible, while taste-masking and controlled release are also present.
Such high API loading and entrapment is made possible because the Optimμm fabrication method can process suspensions and high viscosity mixtures while shearing the molten stream to less than 1/5 of the nozzle orifice before layered droplet breakup and hardening.
Here, we will look closely at specific formulation examples representing some of the highlighted techniques for producing controlled-release powder formulations. Approaches detailed include hot-melt extrusion paired with spinning disk atomization, fluidized bed coating, ion exchange resins, and precision particle fabrication technology. Depending on the steps involved, either an immediate-, delayed-, or extended-release formulation is achieved.
4.1 AzithromycinThis example outlines manufacture of immediate-release microspheres containing azithromycin via hot melt extrusion and spinning disk atomization (Appel L. et al., 2005).
A mixture consisting of 370 g microcrystalline wax, 90 g Pluronic® F87, and 90 g stearyl alcohol was added to a glass beaker in a water bath, and was melted over 60 minutes at 93 °C. Following melting, 450 g azithromycin dihydrate was added such that the anticipated batch size was 1.0 kg with a theoretical drug loading of 45 %. This mixture was mixed for an additional 25 minutes.
Using a gear pump, the molten azithromycin solution was transferred at 250 mL/min onto the center of a spinning disk atomizer, which was rotating at 8000 rpm heated to 100 °C. The microparticles solidified mid-air via convection, were collected in a lined vessel, and determined to have a mean diameter of 190 μm.
Following manufacture, the powder was dissolution tested with a United States Pharmacopeia (USP) Type II apparatus (Fig. 4), which demonstrates the immediate-release nature of the un-coated formulation.

Dissolution of uncoated azithromycin microspheres created with spinning disk atomization. Paddle speed was 50 rpm in a 37 °C vessel with simulated gastric fluid. Image created from tabulated dissolution data in US 2005/0152982 A1 (Appel L. et al., 2005).
This example outlines manufacture of delayed-release microcapsules containing cetirizine via hot melt extrusion, spinning disk atomization, then fluidized bed coating. (Appel L. et al., 2012) The manufacture is largely the same as the previous example, with the exception of adding a second coating step to make delayed-release microcapsules.
A mixture consisting of 750 g Compritol® 888 and 62.5 g Pluronic® F127 was added to sealed stainless steel tank, and was melted over 40 minutes at 90 °C with heating fluid circulating through the tank’s jacket. The molten solution was stirred at 75 rpm for 5 minutes. Following melting, 250 g cetirizine and 187.5 g of croscarmellose sodium was added such that the anticipated batch size was 1.25 kg with a theoretical drug loading of 20 %. This mixture was stirred for an additional 5 minutes with a high speed homogenizer.
Using nitrogen, the tank was pressurized to 103 kPa to transfer the molten cetirizine solution at 145 g/min onto the center of a spinning disk atomizer, which was rotating at 5500 rpm heated to 90 °C. The microparticles solidified mid-air via convection, were collected in a lined vessel.
The cetirizine-containing microparticles were then coated with a 15 % solution of Surelease® E77050 for 150 minutes in a Glatt GPCG-1 fluidized bed coater equipped with a Würster column set to 15 mm. Circulating air (~17.9 L/s) and bed temperatures were kept at an average of 63 °C and 46 °C, respectively, while the coating was applied between 3.8 and 7.4 g/min at 220 kPa.
Following manufacture, the powder was dissolution tested with a United States Pharmacopeia (USP) Type II apparatus (Fig. 5), which demonstrates the delayed-release nature of the coated formulation.
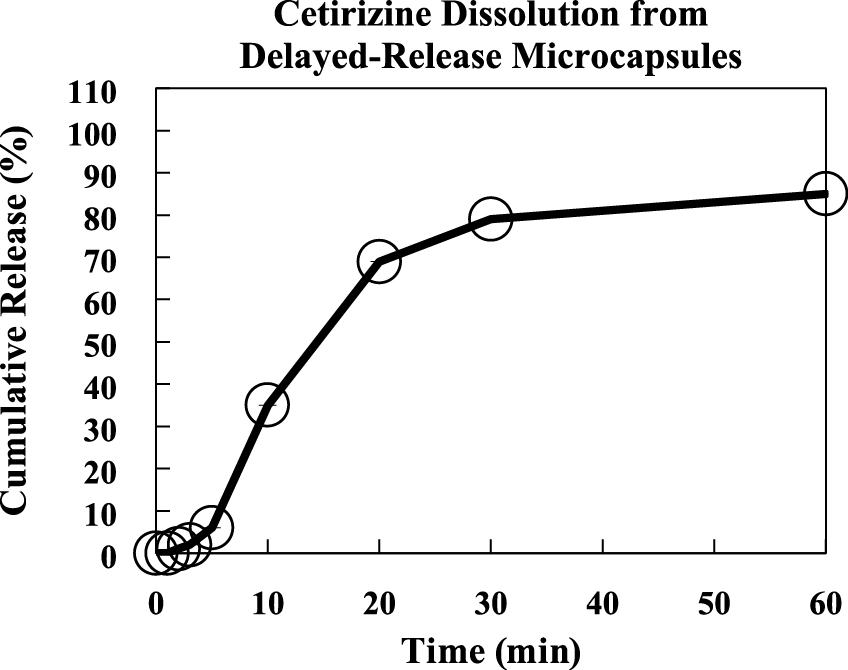
Dissolution of coated cetirizine microcapsules created with spinning disk atomization and Würster coating. Paddle speed was 50 rpm in a 37 °C vessel with simulated mouth buffer. Image created from tabulated dissolution data in US 8,236,349 B2 (Appel L. et al., 2012).
This example outlines manufacture of extended-release microcapsules containing potassium chloride via coacervation, fluidized bed coating (Powell T.C., 1995).
A mixture consisting of 567 kg of potassium chloride crystals between 300 μm and 600 μm in diameter were first mixed with 3312 L of cyclohexane, 83.5 kg of ethylcellulose, and 58 kg of polyethylene. The contents were then heated under agitation until all of the components were in solution. The solution was then cooled to achieve initial coating of the potassium chloride crystals, filtered under vacuum, then dried in a fluidized bed, resulting in single-layer microcapsules less than 850 μm in diameter. A second coating is applied in a fluidized bed coater with a solution consisting of 6.9 kg of hydroxypropylcellulose in 35 L purified water, such that the anticipated batch size was 715 kg with a theoretical drug loading of 79 %.
Following manufacture, the powder was dissolution tested with a United States Pharmacopeia (USP) Type II apparatus (Fig. 6), which demonstrates the extended-release nature of the dual-coating formulation.

Dissolution of single-coated potassium chloride microcapsules created with ion-exchange resins. Paddle speed was 100 rpm in a 37 °C vessel with purified water. Image created from tabulated dissolution data in US 5,422,122 (Powell T.C., 1995).
This example outlines manufacture of immediate-release microparticles containing dextromethmorphan via ion exchange resins (Hirsh J. et al., 2005).
First, 1.0 kg of an ion exchange resin, Amberlite® IRP69, was added to 4.75 L of deionized water heated to 90 °C, and was mixed well before adding 300 g of dextromethmorphan HBr. The solution was allowed to mix for 120 minutes to facilitate binding. Next, the resin particles were isolated via vacuum filtration, washed with 10 L of heated deionized water, then re-suspended in another 3 L of heated deionized water. An additional 300 g of dextromethmorphan HBr was then added to the 3 L volume under mixing, which continued for an additional 120 minutes.
Following second stage binding, the reaction mixture was cooled overnight, filtered, washed three times with 10 L volumes of heated deionized water, and dried at 45 °C until substantially free of water as determined by a moisture analyzer. The resulting microparticles had a drug load of approximately 31 %.
Following manufacture, the powder was dissolution tested with a United States Pharmacopeia (USP) Type II apparatus (Fig. 7), which demonstrates the immediate-release nature of the un-coated formulation.

Dissolution of uncoated dextromethmorphan microparticles created with ion exchange resins. Paddle speed was 50 rpm in a 37 °C vessel with 0.1 N HCl. At 1 hour, the buffer pH was increased to 6.8. Image created from tabulated dissolution data in US 2005/0181050 A1 (Hirsh J. et al., 2005).
This example outlines manufacture of an extended-release microparticle formulation containing methylphenidate via ion exchange resins and fluidized bed coating (Mehta K. et al., 2012). The manufacture is largely the same as the previous example, with the exception of adding second and third coating steps to make extended-release microcapsules. The entire formulation, however, uses single-coated, dual-coated, and drug-free diluent particles to achieve the extended release profile.
First, 3.1 kg of methylphenidate HCl was solubilized in 80 L of purified water with continuous mixing, before adding 7.7 kg of Amberlite® IRP69. This solution was allowed to mix 60 minutes before being filtered, rinsed, and dried until water content was between 3 % and 7 %. The methylphenidate-resin complex was milled and sieved such that particles under 410 μm were isolated for the next step.
In a secondary containment system, 657 g of Kollidon® K30 was dissolved in 2.7 L of purified water, to which the methylphenidate resin complexes were added under mixing until the Kollidon® solution gained nearly 8 % in weight. The slurry was dried until the moisture content was between 15 % and 25 %, milled and sieved a second time to yield particles under 410 μm, dried until water content was between 3 % and 7 %, then milled and sieved a third time to yield particles under 410 μm. The resulting product were single-coated particles.
Dual coated particles were manufactured by applying a second coat to the methylphenidate resin complexes detailed above. A solution consisting of 2.1 kg of purified water, 56 g triacetin, and 3.4 kg Kollicoat® SR30D was prepared and fed to a fluid bed processor with Würster column to coat approximately 3.9 kg of methylphenidate particles from the previous coating step. This was performed until the particles achieved 30 % weight gain. As a last step, the dual-coated methylphenidate ion exchange complex was dried for 5 hours at 60 °C before being passed for a fourth time through a 410 μm mesh screen.
The third component, diluent microparticles, were prepared by mixing 125 g Poloxamer® 188, 34.4 kg sugar, 965 g sodium citrate, 1.3 kg anhydrous citric acid, 500 g sodium benzoate, and 200 g sucralose to 1.8 L purified water in a high shear granulator. The granules were dried until the water content was below 1.5 %, then milled and sieved through an 850 μm mesh screen.
The entire extended release composition consisted of starch, xanthan gum, talc, flavor, and silicon dioxide such that the diluent granules, single-coated granules, and dual-coated granules, approximated 75 %, 1.7 %, and 9.9 % of the total dry weight, respectively.
Following manufacture, the above components were reconstituted to an equivalent of 60 mg methylphenidate in water, and administered to healthy adults. Mean plasma concentration of methylphenidate was compared to adults administered two 30 mg dose of the RLD product (Fig. 8), which demonstrates the extended-release nature of the dual-coated formulation.

Pharmacokinetics of a suspension of single-coated, dual-coated, and diluent methylphenidate microcapsules. Plasma concentration was also compared to administration of two 30 mg RLD tablets. The data demonstrate that the microcapsule suspension maintains plasma concentration of methylphenidate as two separately-dosed tablets would. Image reproduced from US 8,287,903 B2 (Mehta K. et al., 2012).
This example outlines manufacture of taste-masked microspheres containing prednisone via precision particle fabrication (Berkland C. and Singh M., 2014, 2015; Berkland C. et al., 2014). The manufacture is largely the same as the previous example, with the exception of using a dual chamber nozzle for simultaneous coating to yield delayed-release microcapsules.
A mixture consisting of 120 g glyceryl monostearate and 50 g sorbitan monostearate was melted at 95 °C under constant stirring in a jacketed glass reactor under constant mixing. Following, 20 g Eudragit® EPO was added until dispersed. This mixture was allowed to mix for 2 hours, followed by addition of 10 g of prednisone.
The resulting solution was transferred to a 200 mL stainless steel syringe loaded onto a Harvard Apparatus PHD Ultra syringe pump. The syringe was maintained at 80 °C and the contents were injected to primary chamber of a single-barrel stainless steel precision particle fabrication nozzle heated to 80 °C. The flow rate of the prednisone solution was 35 mL/h. such that the anticipated batch size was 200 g with a theoretical prednisone loading of 5.0 %. A third chamber was fed filtered ambient nitrogen at 3.0 L/min to stabilize the droplet breakup. The nozzle was subjected to a frequency of 1.0 kHz with an amplitude of 1.0 Volts, peak-to-peak (Vpp).
The microspheres solidified mid-air via convection and were collected in a stainless steel vessel lined with a 2 mm poly-bag. Resulting particles had a mean size of 180 μm in diameter.
Following manufacture, the powder was dissolution tested with a United States Pharmacopeia (USP) Type II apparatus (Fig. 9) and compared to an RLD syrup, which demonstrates the delayed-release nature of the single-step coated formulation.

(Top) Dissolution of coated prednisone microcapsules created with precision particle fabrication. Paddle speed was 75 rpm in a 37 °C vessel with 0.1 N HCl. Data are compared to the RLD tablet. (Bottom) Microsphere rupture in vitro after pH change.
This example outlines manufacture of extended-release microspheres containing guaifenesin via precision particle fabrication (Berkland C. and Singh M., 2014, 2015; Berkland C. et al., 2014), where a coating step is not included to achieve extended release.
A mixture consisting of 1360 g carnauba wax, 200 g stearic acid, and 20 g ethylcellulose was melted at 95 °C under constant stirring in a stainless steel mixing vessel. After the molten mixture was formed, 640 g of guaifenesin was incorporated until a homogenous solution was formed such that the anticipated batch size was 1.0 kg with a guaifenesin loading was 32 %.
The resulting solution was pressurized with filtered nitrogen and fed at 1.0 kg/h through a mass flow controller, to the primary chamber of a stainless steel 12-barrel precision particle fabrication nozzle heated to 95 °C. A secondary chamber was fed filtered ambient nitrogen at 3.6 L/min to stabilize the droplet breakup. The nozzle was subjected to a frequency of 1.4 kHz with an amplitude of 1.0 Vpp.
The microspheres solidified mid-air via convection and were collected in a stainless steel vessel lined with a 2 mm poly-bag. Resulting particles had a mean size of 150 μm in diameter.
Following manufacture, the powder was dissolution tested with a United States Pharmacopeia (USP) Type II apparatus (Fig. 10) and compared to an RLD tablet, which demonstrates the extended-release nature of the un-coated formulation.

(Top) Dissolution of uncoated guaifenesin microspheres created with precision particle fabrication. Paddle speed was 75 rpm in a 37 °C vessel with 0.1 N HCl. Data are compared to the RLD tablet, where the microsphere formulation has an F2 (similarity factor) of 97.2. (Bottom) Scanning electron micrograph image of guaifenesin microspheres with a mean size of 150 μm in diameter.
This example outlines manufacture of extended-release microspheres containing ibuprofen via precision particle fabrication (Berkland C. and Singh M., 2014, 2015; Berkland C. et al., 2014), where a coating step is not included to achieve extended release.
A mixture consisting of 3150 g carnauba wax, 250 g stearic acid, and 100 g ethylcellulose was melted at 95 °C under constant stirring in a sealed 30 L stainless steel mixing vessel with high shear homogenizer. After the molten mixture was formed, 1500 g of ibuprofen was incorporated until a homogenous solution was formed such that the anticipated batch size was 5.0 kg with an ibuprofen loading was 30 %.
The resulting solution was pressurized with filtered nitrogen and fed at 2.0 kg/h through heat-traced process lines and distributed to five mass flow controllers, feeding the primary chambers of five stainless steel 12-barrel precision particle fabrication nozzles heated to 95 °C. The five secondary nozzle chambers were each fed filtered ambient nitrogen at 5 L/min/nozzle to stabilize the droplet breakup. The nozzles were subjected to a frequency of 2.0 kHz with an amplitude of 0.6 Vpp.
The microspheres solidified mid-air via convection inside an enclosed 825 L jacketed stainless steel vessel chilled with heat transfer fluid to a temperature of 15 °C. Resulting particles had a mean size of 120 μm in diameter.
A similar formulation was also made on a single-barrel precision particle fabrication nozzle that with a composition of 45 % carnauba wax, 25 % stearic acid, and 30 % ibuprofen.
Following manufacture, the powders were dissolution tested with a United States Pharmacopeia (USP) Type II apparatus (Fig. 11) and compared to an RLD tablet, which demonstrates the extended-release nature of the un-coated formulations.

Dissolution of uncoated 12-hour and 24-hour extended-release ibuprofen microspheres created with precision particle fabrication. Paddle speed was 75 rpm in a 37 °C vessel with 0.05 M SLS. Data are compared to the RLD tablet.
This example outlines manufacture of taste-masked microcapsules containing ritonavir via precision particle fabrication (Berkland C. and Singh M., 2014, 2015; Berkland C. et al., 2014). The manufacture is largely the same as the previous example, with the exception of using a dual chamber nozzle for simultaneous coating to yield delayed-release microcapsules.
A mixture consisting of 40 g stearic acid and 10 g Eudragit ® EPO was melted at 95 °C under constant stirring in a stainless steel mixing vessel. This mixture would comprise the microcapsule shell feed.
A second vessel was prepared where 42.5 g of polyethylene glycol 1500 was melted at 80 °C. Following, 7.5 g of ritonavir was incorporated until a homogenous dispersion was formed. This mixture would comprise the microcapsule core feed.
The resulting solutions were each transferred to 50 mL stainless steel syringes loaded onto a Harvard Apparatus PHD Ultra syringe pumps. The syringes were maintained at 80 °C and the contents were simultaneously injected to primary and secondary chambers of a single-barrel stainless steel precision particle fabrication nozzle heated to 80 °C. The flow rates of the prednisone-free shell and prednisone-containing core were set to 30 mL/h and 20 mL/h, respectively, such that the anticipated batch size was 90 g with a theoretical prednisone loading of 6.7 %. A third chamber was fed filtered ambient nitrogen at 2.5 L/min to stabilize the droplet breakup. The nozzle was subjected to a frequency of 1.0 kHz with an amplitude of 1.0 Vpp.
The microcapsules solidified mid-air via convection and were collected in a stainless steel vessel lined with a 2 mm poly-bag. Resulting particles had a mean size of 250 μm in diameter.
Following manufacture, the powder was dissolution tested with a United States Pharmacopeia (USP) Type II apparatus (Fig. 12) and compared to an RLD syrup, which demonstrates the delayed-release nature of the single-step coated formulation.

Dissolution of coated delayed-release ritonavir microcapsules created with precision particle fabrication. Paddle speed was 75 rpm in a 37 °C vessel with 0.1 N HCl for the first 2 minutes, then 0.05 M SLS. Data are compared to the RLD syrup.
Though traditional oral dosage forms such as pills, capsules, caplets, and tablets work for many individuals, a significant fraction of the world’s population suffers from dysphagia, taste sensitivities, or an unwavering avoidance to taking oral medication of any format. As these patients are afflicted with acute or chronic illnesses, sometimes a lack of format flexibility and dosage options limits treatment, and in the worst circumstances, prevents it.
An emergence in manufacturing of controlled-release powders has taken place over the last decade, replacing large tablets with dispersible and dose-flexible alternatives that can be supplied in sachets, sprinkled in food, or suspended in liquid. The methods for making controlled release powders vary, but typically include forming an API/excipient core precursor then coating with one or more controlled-release layers for a finished product. Contemporary techniques incorporate chemical modification and sequestration of the API, prior to secondary coating steps. Next-generation techniques eliminate the need for multiple steps, achieving even coatings while maintaining monodisperse size distributions and high API content at small overall particle size to enhance palatability and mouth-feel. Though the number of powder alternatives that are available for currently approved medications is limited, a rising number of pharmaceutical companies and manufacturing organizations are incorporating controlled-release powder manufacturing to their portfolios to address the growing dosage form problem for paediatric and geriatric patients. In an age where “bigger is better,” for nearly everything in culture, the opposite is unquestionably becoming true for powders, where “less is more” in terms of dosage form size, manufacturing steps, and process chemistry.
particle size (μm)
llength (m)
mmass (kg)
Ppressure (Pa)
ttime (s)
Δttime duration
Ttemperature (K)
Vvolume (L)
μgas (Pa s)
ρgdensity (kg m−3)
Vppvolts, peak-to-peak
Martin G. Teresk
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/34_2017013_13.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
Martin Teresk, PhD, joined Orbis after completing five years at Gilead Sciences, where he served as a process development chemist. Martin’s expertise resides in designing, developing, and validating robust and commercially viable processes for the manufacturing of active pharmaceutical ingredients. He holds a PhD in Organic Chemistry from University of Texas at Austin and a BS in Chemistry from Purdue.
Cory J. Berkland
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/34_2017013_14.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
Cory Berkland, PhD, has been developing microencapsulation and drug delivery capabilities for more than a decade. Cory has a PhD in Chemical and Biomolecular Engineering from the University of Illinois, Urbana-Champaign, where he co-invented and developed the Orbis technology. Cory is a Professor of Pharmaceutical Chemistry and Chemical and Petroleum Engineering at The University of Kansas.
Nathan H. Dormer
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/34_2017013_15.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
Nathan Dormer, PhD, leads the formulation team at Orbis Biosciences. He has more than 8 years of experience developing a variety of controlled-release solutions using microsphere techniques. He received his BS in Chemical Engineering from The University of Kansas before completing his PhD in Bioengineering from The University of Kansas with NIH-sponsored training in drug delivery and protein stability. He has authored a number of publications and book chapters relating to microspheres.