近年のシーケンシング技術の進展により多量のゲノムデータが得られるようになり,同時に遺伝的多型と表現型との関連を解析する Genome Wide Association Study (GWAS) も盛んに行われている.これらの情報を誰もが簡単な操作で閲覧することを可能にし,また公開することでデータの利活用を促進することを目的としてウェブブラウザ TASUKE の開発を行った.TASUKE は多サンプルのゲノムリシーケンスデータから得られる多型情報や GWAS の解析結果を幅広い解像度で情報量豊かに表示する.数百サンプル以上の一塩基多型や挿入欠失といった多型情報およびそれらの遺伝子への効果情報,デプス情報,遺伝子アノテーション情報,GWAS のマンハッタンプロット等を並列的に表示し,1 塩基対の解像度から最大 2,000 万塩基対までの広い範囲の情報をスムーズに閲覧することが可能である.これにより,各サンプル間の関係を遺伝子から広域なゲノム領域のレベルで理解することができ,また,GWAS 結果から表現型多型の原因遺伝子の候補を探索することが容易になる.さらに,ゲノムデータの閲覧から得られた情報を実験で活用するため,任意の多型サイトや領域に対して,多型情報を考慮したプライマーの設計や系統樹作成等のさまざまな機能が実装されている.本 TASUKE システムの概要を活用例と合わせて紹介する.
農業食品分野を含めあらゆる生命科学研究において,全遺伝情報であるゲノム情報の活用が進んでいる.ゲノム配列を高速大量に決定することが可能なシーケンシング技術の進展により,多くの農作物種において大規模なゲノム情報を利活用することが可能となってきた.ゲノムワイドな品種間の遺伝的な差異(多型)情報は,表現型形質の責任遺伝子領域を調べる研究や,特定の形質の責任遺伝子領域を交配育種で導入する際に用いる遺伝マーカーの設計などに有用である.農業分野において,ゲノム多型情報を利用した研究では,イネ,トウモロコシ,ムギ類といった主要な農作物種では大規模な国際プロジェクトにより 1,000 以上の品種サンプルのゲノム情報が取得されているが,その他多くの農作物種では数十~数百品種程度のゲノムデータが取得されている.
さまざまな種でゲノム情報を用いた研究が加速している背景の一つに,ロングリード型といわれるシーケンサーの飛躍的な発展により高品質な参照ゲノム配列が構築できるようになった状況がある.ゲノム多型解析に主に用いられる第二世代シーケンサーといわれるショートリード型のシーケンサーが1配列あたり数百塩基までの解読を並列的に超大量に行うのに対して,PacBio 社や Oxford Nanopore Technologies 社の第三世代のロングリード型シーケンサーは1配列あたり数万~最大で数十万塩基を超えるようなリード配列を得ることができる.このようなデータはゲノム多型解析の基盤として必要な参照ゲノム配列を構築する上で極めて有効である.ロングリード型シーケンサーを活用することで,ゲノム配列構築で最初におこなうアセンブルにおいて,非常に長い contig 配列を構築することが可能である.また,contig 配列間の位置関係を調べて整列させるスキャフォールディングの手法についても,より遠距離な位置関係の情報を物理的な近接情報から得る Hi-C や Omni-C といった手法,DNA 鎖の制限酵素サイトを蛍光標識し,その位置を読み取るオプティカルゲノムマッピングなどの新しい技術により,さまざまな農作物種において染色体レベルに繋がった高品質な参照ゲノム配列が短期間で構築できるようになっている.高品質な参照ゲノム配列と数百品種程度のゲノム多型情報といったデータセットが盛んに産生されている状況である.
このようにゲノム研究が著しく進展する状況でデータの取得と並んで重要となるのが,ゲノムデータを最大限に利活用するための基盤技術となるゲノム情報を可視化するためのツールの整備である.農作物種のゲノムサイズは数億塩基対から大きい種では百億塩基対を超えるほどの大きさがある.このゲノム中には何らかの機能タンパク質をコードする遺伝子が数万個以上存在する.品種間のゲノム多型は各農作物種のゲノムサイズや比較する品種によるが,数万から多い場合には百万か所以上も得られる.このような大規模なデータは通常のパソコンのソフトウェアでは扱うことが難しく,専用のソフトウェアが必要である.ゲノム情報に特化した「ゲノムブラウザ」と総称されるソフトウェアは目的に応じてさまざまに開発されているが,多サンプルのゲノム多型情報の可視化に特に主眼を置いたブラウザは存在していなかった.我々は,大規模なゲノム多型情報をより多くの研究者が利活用するための基盤となるゲノムブラウザ TASUKE を開発した(Kumagai et al. 2013).本稿では最新のバージョンである TASUKE+(Kumagai et al. 2019)の機能紹介とオミクス情報基盤の中での活用例を紹介する.
全遺伝情報を対象としたゲノムや全遺伝子発現情報であるトランスクリプトームといった膨大なデータからなるオミクス情報を活用するために,さまざまな情報基盤が開発されている.これらの研究では,データベース化とそのデータを見せるためのブラウザ機能と検索機能を提供することにより,非常に膨大なデータから利用者がそれぞれの目的に応じたデータを見つけ,利用することを可能にしている.モデル植物であるイネは 2004 年に高品質な参照ゲノム配列が日本を中心とした国際プロジェクト the International Rice Genome Sequencing Project (IRGSP)により決定され,シロイヌナズナと並び最もオミクス情報研究が進んでいる植物種である.参照ゲノム配列情報の活用において最も重要なものは遺伝子アノテーション情報の整備である.イネでは,遺伝子アノテーション情報の高精度化のための人の手によるキュレーションが継続的に行われ(Itoh et al. 2007),農研機構が開発,運用する Rice Annotation Project Database (RAP-DB)(Sakai et al. 2013,Kawahara et al. 2013)から公開されている.また別に,米国のミシガン州立大学(MSU)の Rice Genome Annotation Project Database (Yuan et al. 2003) のイネ遺伝子アノテーションも存在するが,現在 RAP-DB では両者を統合したデータが提供されている.RAP-DB には参照ゲノムのブラウジング,遺伝子やマーカー情報などの検索機能,データダウンロード機能をはじめさまざまな機能が盛り込まれ,イネ研究の中心として機能するゲノムデータベースとなっている.その他,イネの各種オミクス情報を整備したデータベースも多数整備されており,RNA-Seq データによる遺伝子の転写プロファイルを表示するための TENOR,共発現遺伝子ネットワーク情報を提供する RiceFREND,圃場サンプルの時系列での遺伝子発現プロファイルを提供する RiceXPro といったデータベースが農研機構で開発,提供されている(Itoh et al. 2018).少々専門的な話になるが,ゲノム配列をベースとしたデータベースを構築可視化するためのツールとして,Generic Genome Browser (GBrowse) や JBrowse がある(Stein et al. 2002, Buels et al. 2016).これらは任意のゲノム配列をベースとしてデータベースを構築可視化することができるように開発されたものであり,さまざまな生物種のゲノム情報を可視化したデータベースで活用されている.このように特定のプロジェクトのために専用設計したシステムではなく,幅広い種のゲノム多型情報の利活用への貢献を目指して汎用的な利用を想定して開発することにより,波及効果の大きな研究開発となることが期待される.
ゲノム多型を対象とした研究では,次世代シーケンサー(Next Generation Sequencer : NGS)から出力されたリード情報(fastqファイル)を低クオリティ塩基やシーケンサーにかけるために必要なアダプター配列などを除去する前処理を行ったのち,参照ゲノム配列へマッピングしアライメントデータ(Sequence Alignment / Map format : SAMファイル)および圧縮した binary 形式の BAM ファイルを作る.この情報から各サイトの遺伝子型を推測し多型情報ファイル(Variant Call Format : VCFファイル)を出力する.研究論文を出版する際には,実験データの公開が義務付けられているジャーナルが多いが,NGS データについては生データであるリードデータがこの対象となる.そのため,DNA 多型情報を一般に広く活用してもらうことを目的とした研究以外は,fastq データのみが公共データベースから公開されているケースが圧倒的に多い.したがって,このような研究の多型情報を参照したい場合には,この fastq データ(数十 G バイト以上もある)を取得して自分自身で再度情報解析を行う必要がある.そのため,情報解析技術を持たない研究者にはせっかくのデータが容易には活用できない状況である.また一方で,多型情報を格納した VCF ファイルを公開している場合もあるが,ゲノム全体にわたる多型情報はあまりに膨大であり,活用するためにはやはり情報解析スキルが必要となる.VCF ファイルなどを扱うことができる Integrative Genomics Viewer (IGV) (Robinson et al. 2011)といった優れた可視化ソフトウェアもあり,比較的小規模なデータセットに対しては非常に有用であるが,大規模なサンプル数の多型情報を広域的に閲覧することは難しい.
ゲノム情報から何を得たいか?を考えた時に,その答えは研究者や研究テーマによるということになる.そのため,TASUKE の開発では,できる限り広い領域の研究者が,欲しい情報を得ることができるようなシステムや機能を考えた.TASUKE の名前の由来は「多くを助ける」という意味を込めた.TASUKE は数百サンプル規模の大規模なゲノムリシーケンシングデータをスケーラブルかつ快適にブラウンジングするために設計された(Kumagai et al. 2013).2019 年に公開したアップデート版である TASUKE+(以降TASUKEと表記)は,それまでのゲノム多型を表示する機能に加えて,ゲノムワイド関連解析(Genome Wide Association Study:GWAS)の結果データを表示する機能を加えた点が最も大きな特徴である(図1,2)(Kumagai et al. 2019).ユーザーインターフェースはウェブブラウザを用いたアプリケーションとして HTML5 で開発することで利用者が最も簡単にアクセスできる形式にしており,バックエンドとして大規模データの効率的なマネージメントと高速な検索が可能な MySQL データベースを用いている.これらにより,多数のサンプルからなる大規模なゲノムデータを簡単かつ高速に閲覧することを可能としている.ゲノム上の各ポジションにおける一塩基多型(SNP),挿入欠失(InDel)を1 塩基レベルから最大 100 kb までのブロック単位の頻度情報として表示することができ,ウィンドウ位置の移動やサンプルの縦方向のスクロールに対して高速に表示することが可能となっている.また,読み厚(デプス)情報(以下に詳述)を合わせて背景に表示することによって,NGS データから得られる情報を最大限に活用している.1ブロック 100 kb の解像度で 200 ブロックを表示することで,最大 20 Mb の領域を表示することが可能であり,染色体の広い領域にわたる情報を俯瞰することができる点が優れた特徴の1つである.さらに,特定の遺伝子領域の分子系統樹作成,任意の多型サイトや領域を対象とした PCRプライマー設計,塩基配列情報をクエリーとした BLAST 検索といった機能を持つ(図 3).また,搭載されている配列データや多型情報のリストを,標準的な fasta 形式やテキスト形式で加工・出力する便利な機能を有する.



TASUKE は柔軟なデータ連携を可能にするため,外部サイトとの双方向性の連携を容易にする機能を持たせている.特定の領域や遺伝子を表示させるために,URL にゲノム上の位置情報や,遺伝子 ID を記載することにより表示領域を指定することができる.また,TASUKE で表示するアノテーション情報に外部サイトへのリンクを貼ることができる.例えば,TASUKE の遺伝子アノテーショントラックの ID をクリックすることで,RAP-DB の詳細な遺伝子アノテーション情報ページに飛ぶことができる.これらの機能を用いて既存のオミクスデータベースとの連携を図ることにより,両者の利用効果が相乗的に高まることが期待される.
TASUKE では多型の表示形式を複数タイプ選ぶことができる.ブロックウィンドウごとの頻度か 1 bp 単位の絶対位置か,さらに,絶対位置での表示は ATGC の塩基ごとでの色分け,あるいは遺伝子型がホモ(2つの対立遺伝子の両方が非リファレンス型)か,ヘテロ(片方の対立遺伝子が非リファレンス型)かを表示することができる.また SnpEff モードでは各多型が遺伝子機能にどのような影響を与えるか,例えば,機能を壊すような可能性のあるドラスティックな変異をアミノ酸配列にもたらしているかといった影響の程度のカテゴリーを表示することができる(Cingolani et al. 2012).TASUKE では NGS データから得られる情報をさらに最大限に活かすことを目指した.すなわち,リードの読み厚(デプス)の情報である.デプスとは,リードが参照ゲノム配列上の塩基ポジションごとに何本張り付いているのかという情報である.デプス情報から得られるものは,多型として VCF ファイルへコールされていない情報である.NGS から得られるリード情報は全ゲノムに均一に得られるわけではなく,デプスにはムラがある.ランダムに生じるムラや,GC / AT リッチなどシーケンスが読まれにくい領域が存在しており,そういった領域ではデプスが薄かったり,まったく読めていない場合も多々ある.そういった場合には,VCF ファイルの情報のみでは本当に変異がないのか,あるいは存在するが検出できていないのかが判然としない.デプス情報を表示することにより,リードがあるのかないのかが判ることは有益である.近年は複数個体の情報を合わせた多型コールでマルチサンプル VCF を作ることが増えており,影響は少なくなってきてはいるものの十分な注意が必要な点である.また,ショートリードデータからコールできない多型として,比較的大きな欠失やコピー数多型情報がある.これらの情報は多型情報ファイルのみからは得ることが難しいが,リードの張り付いているアライメント情報から観察できることがある.例えば,TASUKE の画面上で,欠失領域(デプスが0のサイト)は黄色でハイライトして表示される.また,コピー数多型がある領域は明らかに周囲の領域よりも非連続的にデプスが厚くなっている.このように,アライメントの情報を集計したデプス情報を表示することにより,これらの大きな挿入欠失やコピー数多型といった情報もユーザーは TASUKE から読み取ることができる(図1).今後これらの多型についても,ロングリードデータの活用や,多型検出ソフトウェアのアルゴリズムの向上等により精度良く検出できるようになることが期待される.
農畜産物種におけるゲノムリシーケンス解析の主要な目的のひとつに,さまざまな重要な表現型形質の変異をもたらす遺伝的な変異を探ることがある.今日,このための一般的な手法として GWAS がある.GWAS は多数の個体の表現型情報とゲノムワイドな多型情報をもちいて統計的に相関関係を探ることにより,表現型形質の責任遺伝子領域を探索する手法である.ゲノムリシーケンシング解析では非常に多数の多型情報が検出されるが,GWAS ではこれらの1サイトごとについて仮説検定結果である確率(p 値) をもとめる.この p 値が低い多型あるいはその周辺に原因となる変異が存在すると期待される.言うまでもなく,この GWAS の結果も膨大なデータポイントを有し,GWAS で絞ることのできる候補領域は,サンプル集団によるが通常数十~数百 kb にわたるため,結果データの閲覧は簡単ではない.TASUKE は多型情報,遺伝子アノテーション情報,GWAS の結果であるマンハッタンプロットを並列表示することにより,GWAS の結果から表現型の原因となる変異候補を効率的に探索することを可能としている(図 2). GWAS 機能タブからは GWAS の結果であるマンハッタンプロットが全染色体で図示され,ズームイン/アウトができるほか,各プロットをクリックすることで,該当領域へ移動することができる.また,多型サイトをp値の低い順にソートしたリストも表示され,ここには遺伝子情報や上記した各変異の遺伝子機能への影響の情報も表示される.TASUKE により,GWAS の結果と,遺伝子情報,多型の遺伝子に与える影響,さらに上述したような多型情報として検出できていないような大きな挿入欠失などの情報が一元的に得られ,責任遺伝子や変異の候補の探索を効率的に行うことができる.我々は GWAS 研究のデモデータとしてYano et al.(2016)の日本の温帯ジャポニカイネ 176 品種を用いた解析データを TASUKE に搭載して公開している (https://tasuke-plus.dna.affrc.go.jp).ユーザーは表示する品種サンプルをアクセッションマネージャーから選択し,希望の品種のみを表示させることも可能である.アクセッションマネージャーは,品種の起源地やグループといったさまざまなメタ情報を参照することができる便利な機能である.
TASUKEはさまざまな種のゲノム多型研究プロジェクトでの活用を前提に開発されている.農研機構高度分析研究センターで運用している RAP-DB(Sakai et al. 2013, Kawahara et al. 2013)やかんきつ類の Mikan Genome Database (Kawahara et al. 2020)などで TASUKE を活用している.特に RAP-DB はイネのゲノム,遺伝子アノテーション情報,遺伝子発現量を示すトランスクリプトームやオープンクロマチン領域を検出するための ATAC-Seq といったオミクスデータ,また育種に応用可能な遺伝マーカーといったさまざまな情報を JBrowse により閲覧することができるが,これまでにさまざまな研究プロジェクトにおいて決定されたイネのゲノムリシーケンシングデータを再解析集約した 685 品種サンプルの大規模なイネ多型データを搭載した TASUKE を提供している(https://rapdb.dna.affrc.go.jp/).また,農研機構遺伝資源センターでは,イネの遺伝資源として多様性を広くカバーした「世界のイネコアコレクション(WRC)」および「日本在来イネコアコレクション(JRC)」を研究用資材として提供しているが,これらのコレクションのゲノム多型情報も近年,整備され(Tanaka et al. 2020, 2021)RAP-DB から TASUKE を用いて公開されている.このゲノム情報,形質情報,遺伝資源としての配布体制が揃った研究資源の提供は,さまざまな分野での研究を強力に後押ししていくことが期待される.他にも,かずさ DNA 研究所が開発運用するさまざまな植物種のゲノム配列やマーカー情報を集めたポータルサイトである,Plant GARDEN においても多数の植物種の TASUKE が構築され,公開されている(https://plantgarden.jp/).その他の TASUKE についても TASUKE ウェブページにリンクの情報がある.
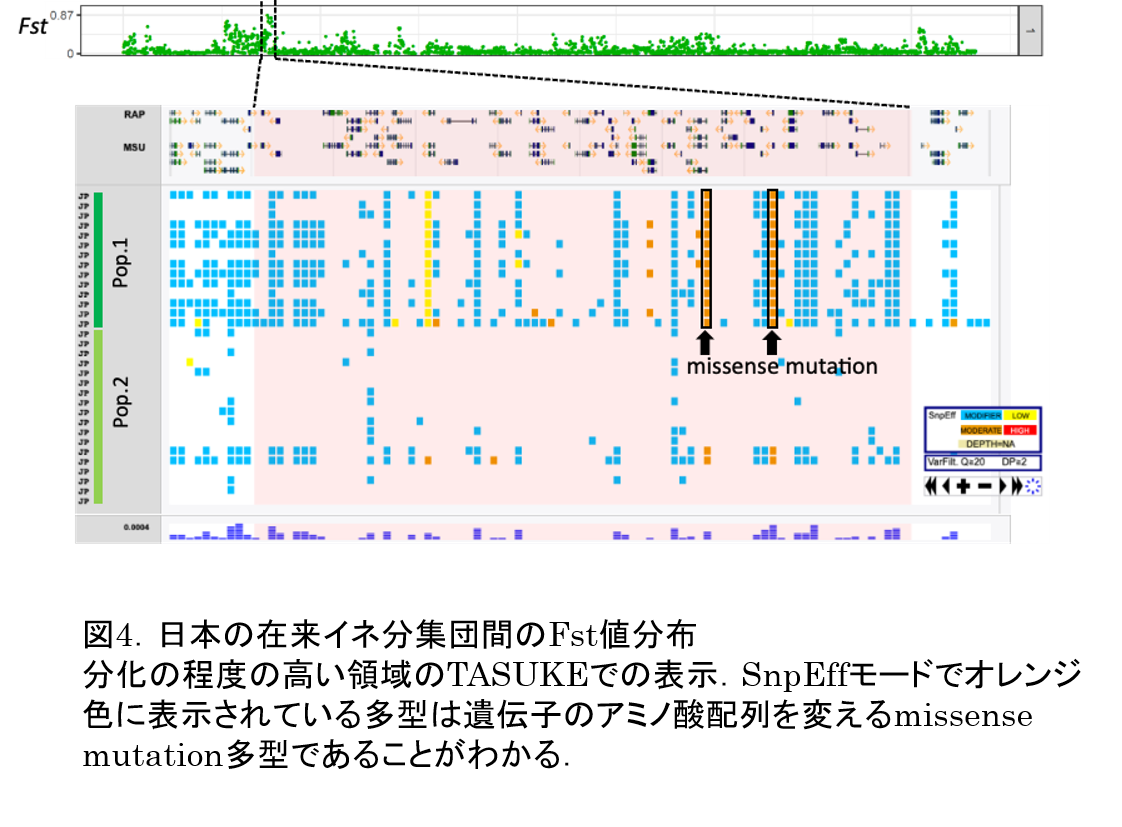
TASUKE を活用する例として GWAS の結果を閲覧して候補遺伝子を探索し,表現型との関連が特定された遺伝子や領域について DNA マーカーを設計し,そのマーカーを増幅するための PCR プライマーを周辺の多型サイトを避けて設計するといった流れが想定される.実際に個別の遺伝子を対象とした研究論文中で集団中の自然変異である多型情報のチェックや,その遺伝子の系統関係を明らかにするために分子系統樹を作成するなど多数活用されている.次に最近の我々の取り組みの中での活用例を紹介する.日本の栽培イネ集団の遺伝的な多様性を前述の日本在来イネコアコレクションの全ゲノムデータを用いて解析した.その中で,日本の在来イネ品種には分集団構造があることがゲノムワイドな多型情報を用いた Admixture 解析により見出された.集団間の分化の程度を測る Fst という統計量を用いて分集団間で対立遺伝子の頻度が分離しているゲノム領域を見いだし,該当領域を TASUKE を用いて探索した(図4).実際に変異のパターンから分化パターンの存在が確認でき,SnpEff モードで表示させると該当領域中に遺伝子の機能への影響を与えている可能性のあるアミノ酸置換型の変異が確認できた.さらに遺伝子アノテーション情報をもとに,これらの遺伝子の詳細な情報を簡単に追うことができる.
TASUKE はエンドユーザーとして,情報解析技術を持たない研究者を想定しているが,TASUKE 自身をセットアップするためには,サーバーの準備や多型データの作成,TASUKE システムのサーバーへのインストールおよび多型データ等のインポートといったコマンドライン入力で行う手順が必要となり,相応の情報解析の知識が必要となる.そこで,この垣根を少しでも下げたいと考え,我々農研機構高度分析研究センターが提供している Galaxy システム Galaxy/NAAC(https://galaxy.dna.aff rc.go.jp/)で TASUKE を利用できるようにしている.Galaxy はグラフィカルユーザインターフェース(GUI)を備えたデータ解析プラットフォームであり,NGS の各種解析をウェブブラウザ上で誰でも非常に簡便に行うことができるシステムである(田中 2020).この Galaxy 上では,ユーザーはリードデータを入力として,前処理,マッピング,遺伝子型のコールといった一連の多型解析を主にマウス操作で行うことができ,さらに得られた多型データを専用の TASUKE に搭載し閲覧することができる.
今後のゲノム研究においては,シーケンシングのコストは下がりつづける予想であり,地球上のあらゆる生物のゲノムを決めてしまおうという流れすらある.農畜産物種においても,ますます大規模なゲノムリシーケンシングが行われ TASUKE システムの活用が進むと考えられる.したがって,データの大規模化への対応が第一の課題である.この点については高速にデータマネージメントや検索をする機能,またデータインポートの効率化を行うための改良が求められる.また,より大規模なデータから効率的に重要な情報を読み取れるようなデータの要約,可視化の新しい方法の開発が課題となっている.第二の課題として,複数の参照ゲノム配列を用いた解析データ,例えばイネではすでに複数の品種で参照ゲノム配列が決定されている(Kumagai et al. 2018)が,ジャポニカとインディカのそれぞれを参照ゲノム配列として行なった多型解析データや,さらに,パンゲノム解析(1つの参照ゲノム配列に限らず,すべての品種が持つゲノムの集合体を解析するための手法)データへの対応が考えられる.オミクス研究全般にいえることだが,これら新しい複雑なデータの対応には従来用いてきたものとは異なる新しい表現方法や効率的なデータの保持方法の開発といったチャレンジングな取り組みが必要となる.また,複数のオミクスデータベース間のさらなる連携や異なる種間のデータの対応づけといった課題を解決していくことが,今後も増え続けるデータを有効に活用していくための情報基盤の開発に求められる.
本TASUKEシステムの開発は農林水産省プロジェクト(GIR1001 および IVG-2001)の中で開発を行なった.本稿中に示した解析データは JSPS 科研費 18H05508,20H05820 の助成を受けたものである.
すべての著者には開示すべき利益相反はない.