
Volume 18
Displaying 1-38 of 38 articles from this issue
- |<
- <
- 1
- >
- >|
Regular Article
-
Article type: Regular Article
2021Volume 18 Pages 1-12
Published: 2021
Released on J-STAGE: February 05, 2021
Advance online publication: January 08, 2021 Download PDF (1476K) Full view HTML
Download PDF (1476K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2021Volume 18 Pages 13-15
Published: 2021
Released on J-STAGE: February 20, 2021
Advance online publication: February 02, 2021Download PDF (297K) Full view HTML
Method and Protocol
-
Article type: Method and Protocol
2021Volume 18 Pages 16-27
Published: 2021
Released on J-STAGE: March 18, 2021
Advance online publication: February 06, 2021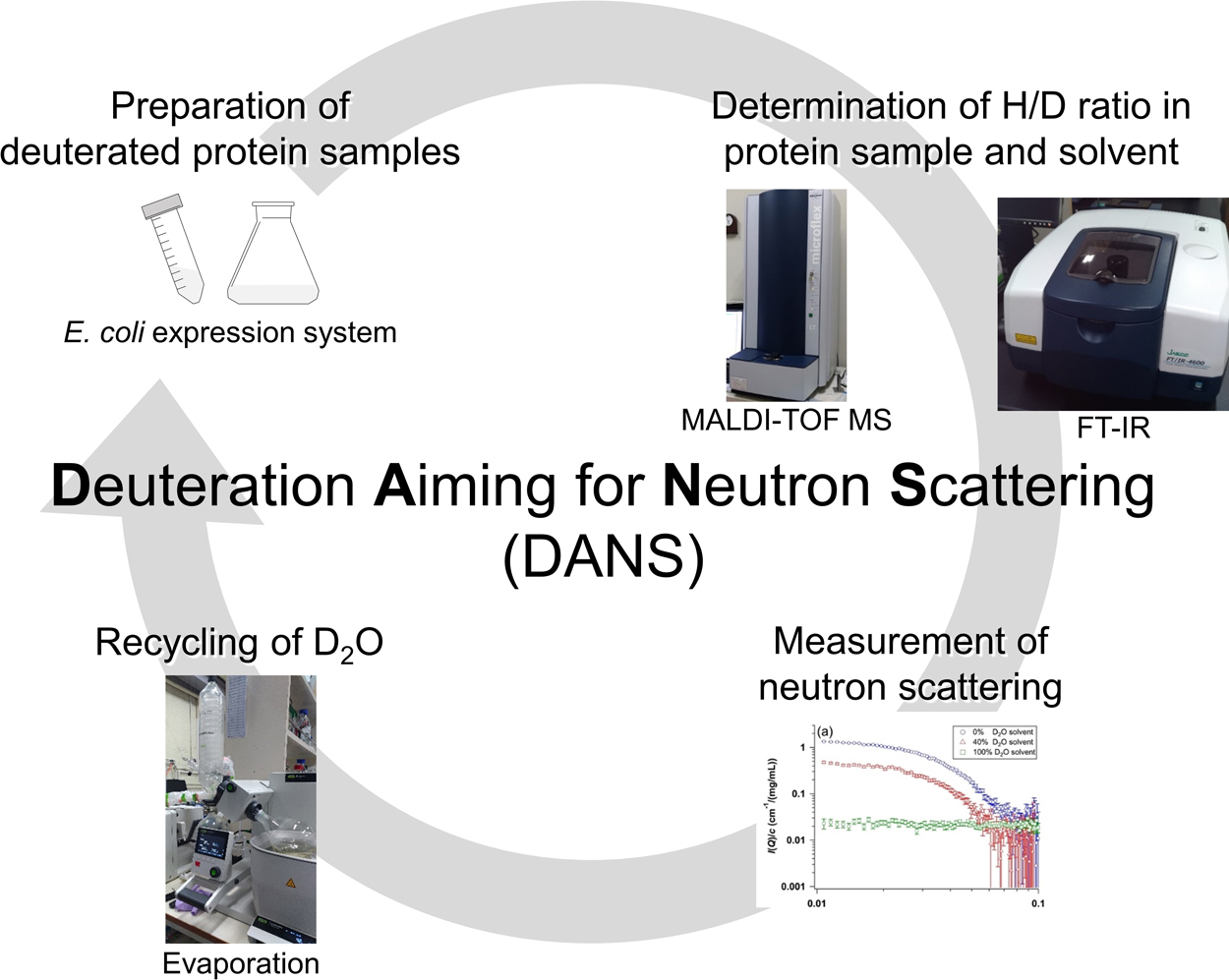 Download PDF (6780K) Full view HTML
Download PDF (6780K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2021Volume 18 Pages 28-39
Published: 2021
Released on J-STAGE: March 18, 2021
Advance online publication: February 10, 2021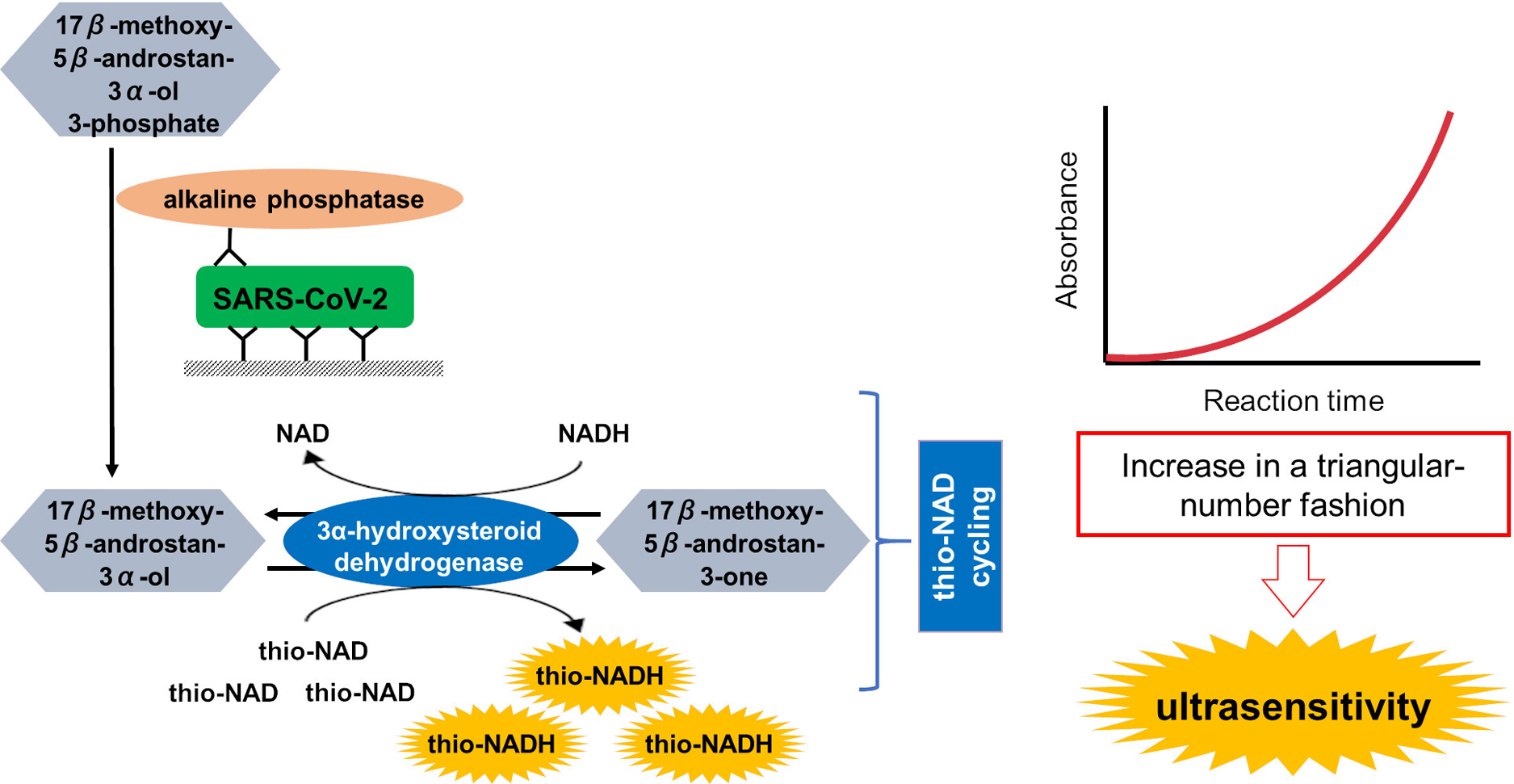 Download PDF (1922K) Full view HTML
Download PDF (1922K) Full view HTML
Regular Article
-
Article type: Regular Article
2021Volume 18 Pages 40-49
Published: 2021
Released on J-STAGE: March 24, 2021
Advance online publication: February 13, 2021 Download PDF (3687K) Full view HTML
Download PDF (3687K) Full view HTML -
Article type: Regular Article
2021Volume 18 Pages 50-59
Published: 2021
Released on J-STAGE: March 24, 2021
Advance online publication: February 18, 2021 Download PDF (3396K) Full view HTML
Download PDF (3396K) Full view HTML
Commentary and Perspective (Invited)
-
Article type: Commentary and Perspective (Invited)
2021Volume 18 Pages 60-66
Published: 2021
Released on J-STAGE: March 27, 2021
Advance online publication: February 18, 2021 Download PDF (1168K) Full view HTML
Download PDF (1168K) Full view HTML
Regular Article
-
Article type: Regular Article
2021Volume 18 Pages 67-77
Published: 2021
Released on J-STAGE: April 13, 2021
Advance online publication: March 17, 2021 Download PDF (3458K) Full view HTML
Download PDF (3458K) Full view HTML -
Article type: Regular Article
2021Volume 18 Pages 78-84
Published: 2021
Released on J-STAGE: April 13, 2021
Advance online publication: March 25, 2021 Download PDF (529K) Full view HTML
Download PDF (529K) Full view HTML -
Article type: Regular Article
2021Volume 18 Pages 85-95
Published: 2021
Released on J-STAGE: April 16, 2021
Advance online publication: April 01, 2021 Download PDF (1830K) Full view HTML
Download PDF (1830K) Full view HTML -
Article type: Regular Article
2021Volume 18 Pages 96-107
Published: 2021
Released on J-STAGE: May 13, 2021
Advance online publication: April 16, 2021 Download PDF (9211K) Full view HTML
Download PDF (9211K) Full view HTML -
Article type: Regular Article
2021Volume 18 Pages 108-115
Published: 2021
Released on J-STAGE: May 13, 2021
Advance online publication: April 16, 2021 Download PDF (3200K) Full view HTML
Download PDF (3200K) Full view HTML
Review Article (Invited)
-
DNA nanotechnology provides an avenue for the construction of programmable dynamic molecular systemsArticle type: Review Article (Invited)
2021Volume 18 Pages 116-126
Published: 2021
Released on J-STAGE: May 26, 2021
Advance online publication: April 27, 2021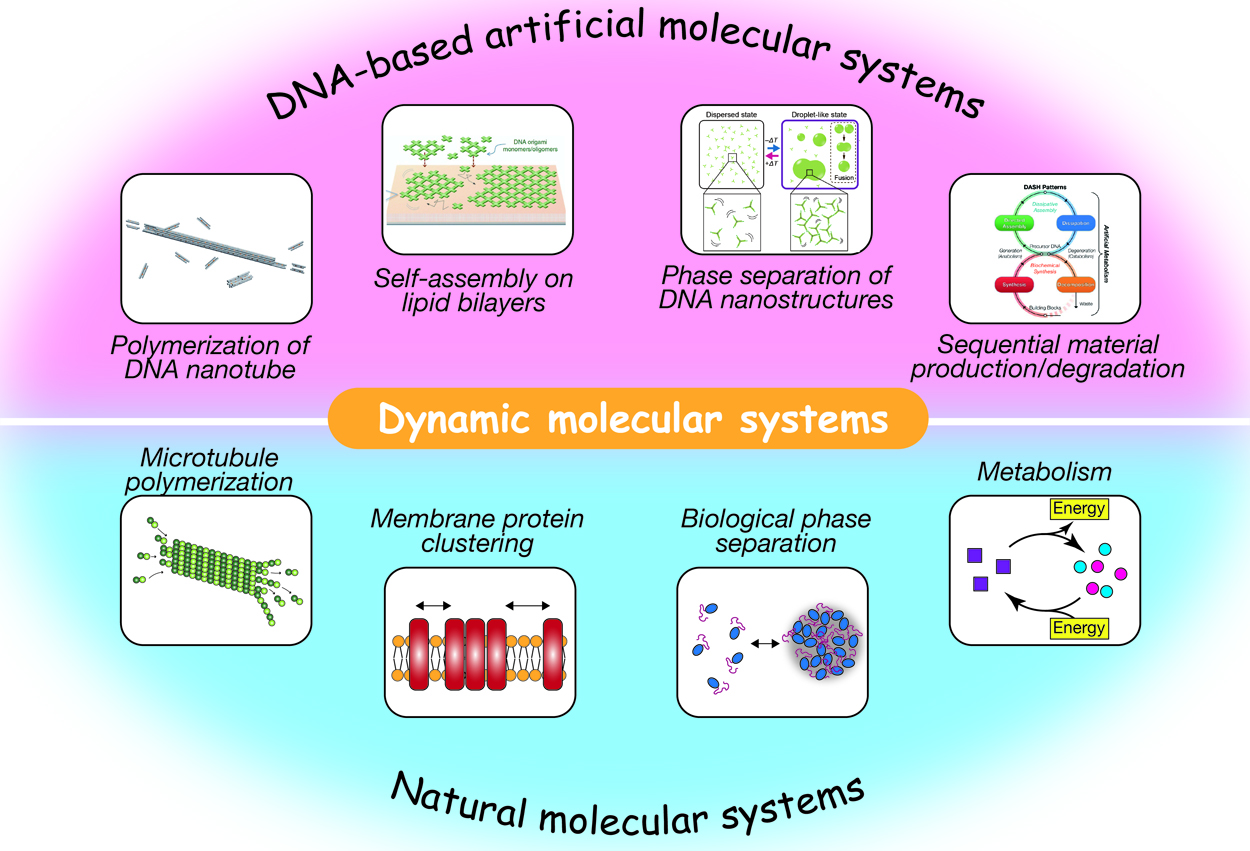 Download PDF (7204K) Full view HTML
Download PDF (7204K) Full view HTML
Commentary and Perspective (Invited)
-
Article type: Commentary and Perspective (Invited)
2021Volume 18 Pages 127-130
Published: 2021
Released on J-STAGE: June 18, 2021
Advance online publication: May 14, 2021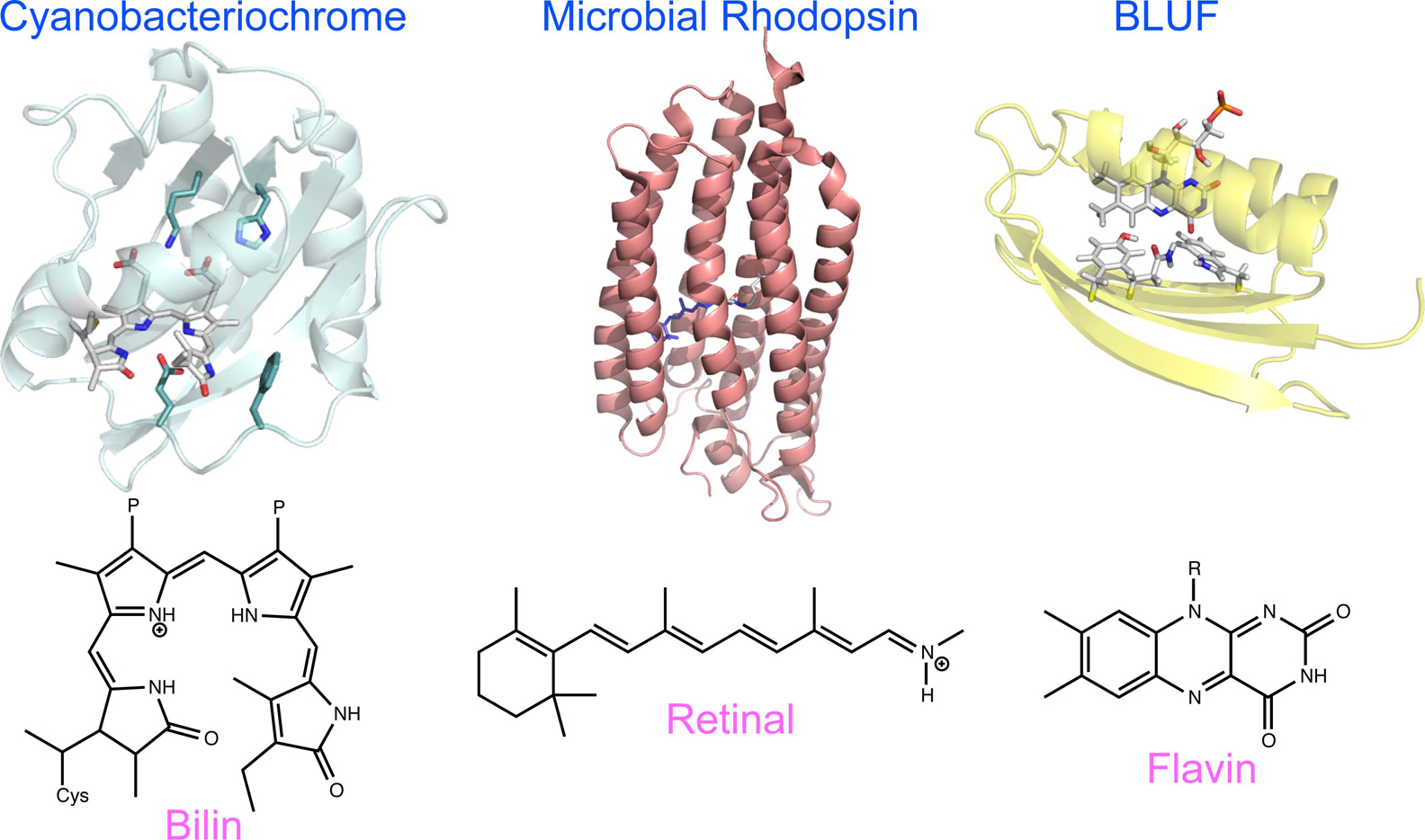 Download PDF (1393K) Full view HTML
Download PDF (1393K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2021Volume 18 Pages 131-144
Published: 2021
Released on J-STAGE: June 18, 2021
Advance online publication: May 15, 2021 Download PDF (1675K) Full view HTML
Download PDF (1675K) Full view HTML
Regular Article
-
Article type: Regular Article
2021Volume 18 Pages 145-158
Published: 2021
Released on J-STAGE: June 18, 2021
Advance online publication: May 21, 2021 Download PDF (1033K) Full view HTML
Download PDF (1033K) Full view HTML -
Article type: Regular Article
2021Volume 18 Pages 159-167
Published: 2021
Released on J-STAGE: June 18, 2021
Advance online publication: May 28, 2021 Download PDF (6948K) Full view HTML
Download PDF (6948K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2021Volume 18 Pages 168-176
Published: 2021
Released on J-STAGE: July 28, 2021
Advance online publication: June 30, 2021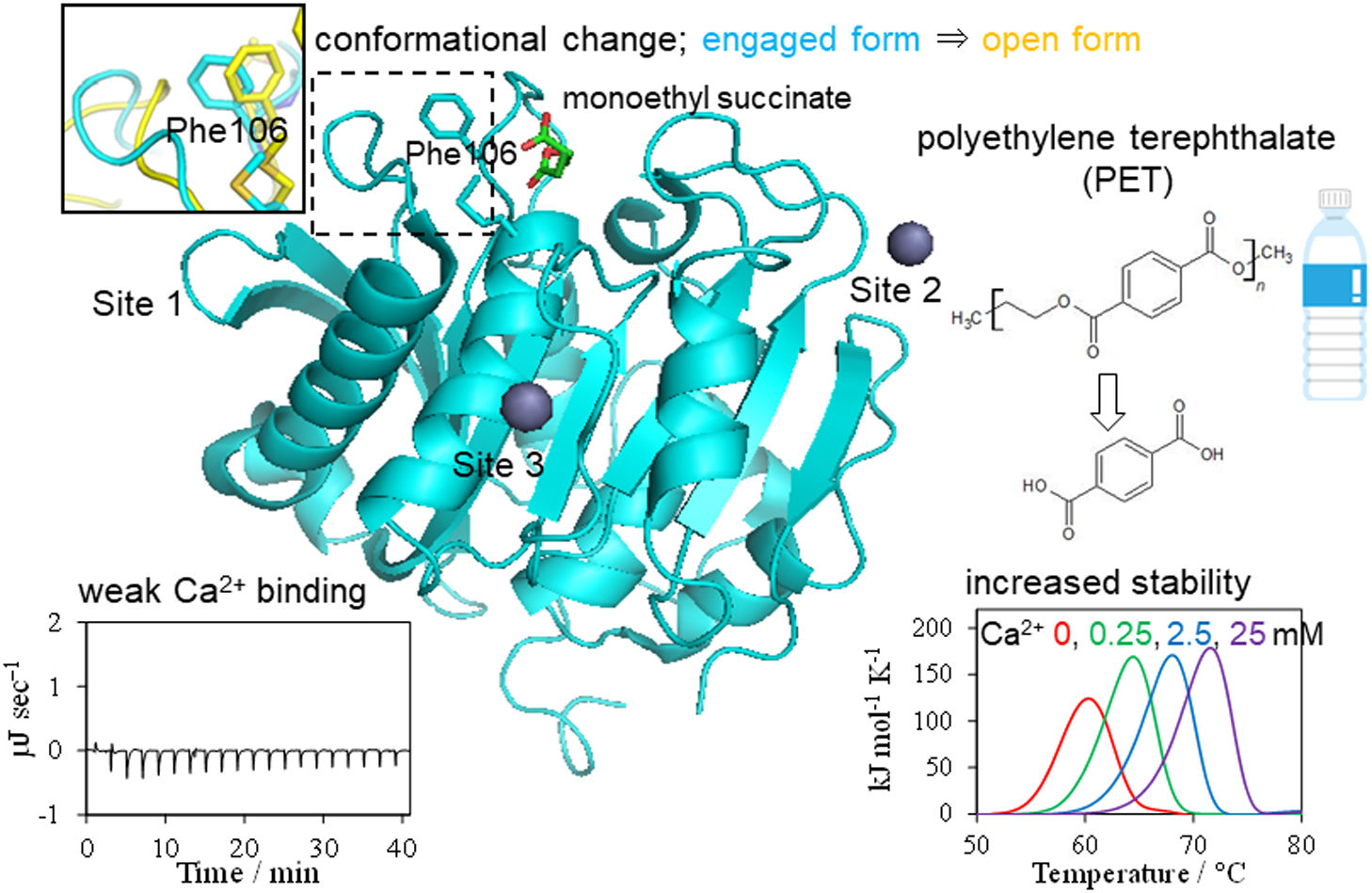 Download PDF (2525K) Full view HTML
Download PDF (2525K) Full view HTML
Regular Article
-
Article type: Regular Article
2021Volume 18 Pages 177-185
Published: 2021
Released on J-STAGE: August 07, 2021
Advance online publication: July 14, 2021 Download PDF (1701K) Full view HTML
Download PDF (1701K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2021Volume 18 Pages 186-195
Published: 2021
Released on J-STAGE: August 19, 2021
Advance online publication: July 16, 2021 Download PDF (6686K) Full view HTML
Download PDF (6686K) Full view HTML
Regular Article
-
Article type: Regular Article
2021Volume 18 Pages 196-214
Published: 2021
Released on J-STAGE: September 01, 2021
Advance online publication: July 30, 2021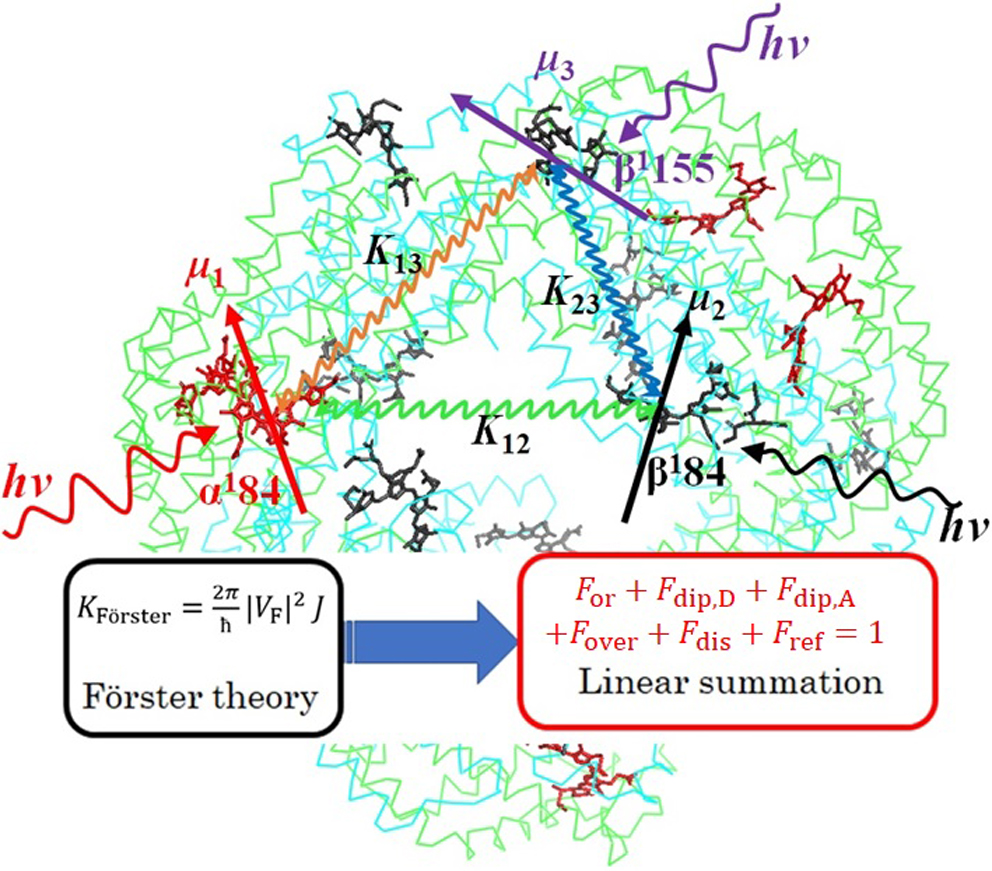 Download PDF (4587K) Full view HTML
Download PDF (4587K) Full view HTML
Note
-
Article type: Note
2021Volume 18 Pages 215-222
Published: 2021
Released on J-STAGE: September 16, 2021
Advance online publication: August 21, 2021 Download PDF (3550K) Full view HTML
Download PDF (3550K) Full view HTML
Editorial
-
Article type: Editorial
2021Volume 18 Pages 223
Published: 2021
Released on J-STAGE: October 01, 2021
Download PDF (185K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2021Volume 18 Pages 224-225
Published: 2021
Released on J-STAGE: October 08, 2021
Advance online publication: October 05, 2021Download PDF (194K) Full view HTML
Review Article (Invited)
-
Current status of structure-based drug repurposing against COVID-19 by targeting SARS-CoV-2 proteinsArticle type: Review Article
2021Volume 18 Pages 226-240
Published: 2021
Released on J-STAGE: October 20, 2021
Advance online publication: October 05, 2021 Download PDF (3506K) Full view HTML
Download PDF (3506K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2021Volume 18 Pages 241-243
Published: 2021
Released on J-STAGE: October 20, 2021
Advance online publication: October 06, 2021Download PDF (1684K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2021Volume 18 Pages 244-253
Published: 2021
Released on J-STAGE: October 26, 2021
Advance online publication: October 15, 2021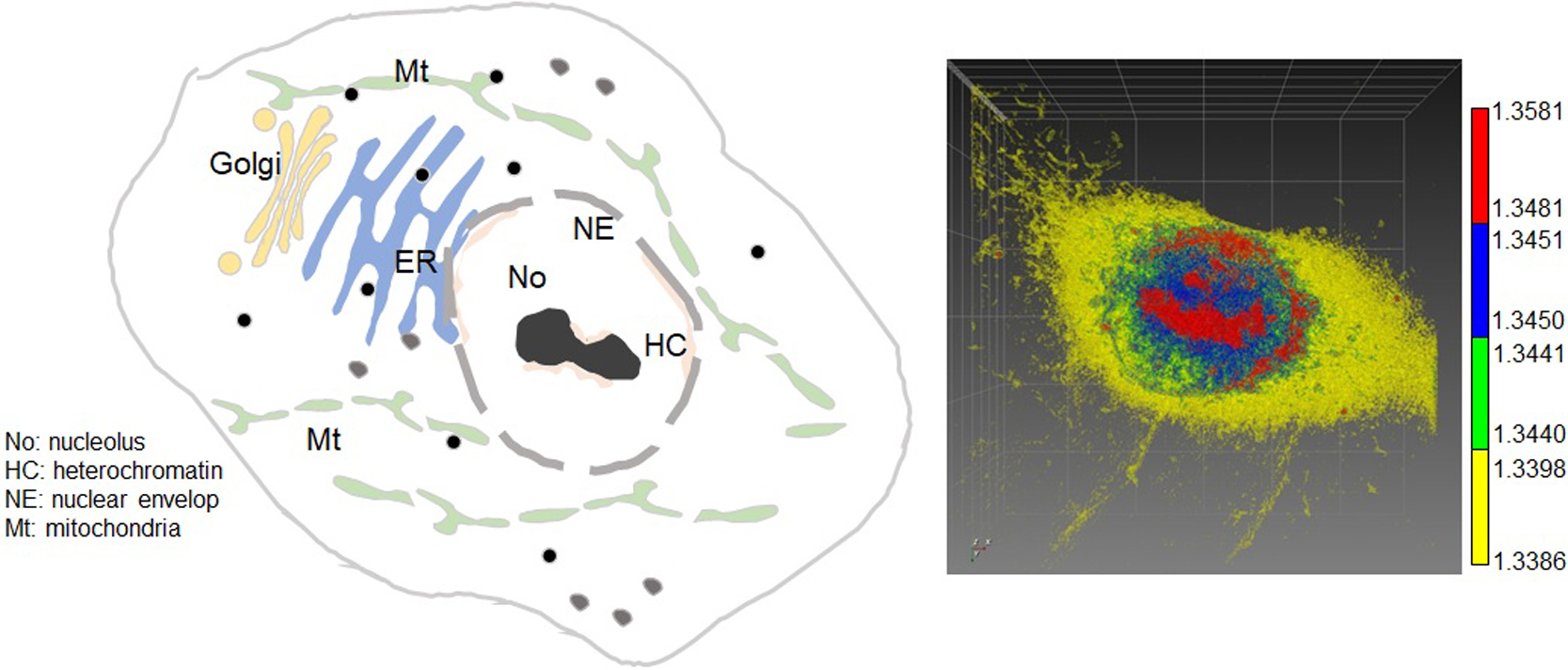 Download PDF (3529K) Full view HTML
Download PDF (3529K) Full view HTML -
Article type: Review Article (Invited)
2021Volume 18 Pages 254-262
Published: 2021
Released on J-STAGE: November 27, 2021
Advance online publication: October 20, 2021 Download PDF (3353K) Full view HTML
Download PDF (3353K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2021Volume 18 Pages 263-264
Published: 2021
Released on J-STAGE: November 27, 2021
Advance online publication: October 21, 2021Download PDF (308K) Full view HTML -
Article type: Commentary and Perspective
2021Volume 18 Pages 265-266
Published: 2021
Released on J-STAGE: November 27, 2021
Advance online publication: October 23, 2021Download PDF (308K) Full view HTML -
Article type: Commentary and Perspective
2021Volume 18 Pages 267-268
Published: 2021
Released on J-STAGE: November 27, 2021
Advance online publication: October 23, 2021Download PDF (320K) Full view HTML -
Article type: Commentary and Perspective
2021Volume 18 Pages 269-273
Published: 2021
Released on J-STAGE: December 02, 2021
Advance online publication: November 18, 2021Download PDF (510K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2021Volume 18 Pages 274-283
Published: 2021
Released on J-STAGE: December 04, 2021
Advance online publication: November 19, 2021 Download PDF (1715K) Full view HTML
Download PDF (1715K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2021Volume 18 Pages 284-288
Published: 2021
Released on J-STAGE: December 11, 2021
Advance online publication: November 23, 2021Download PDF (842K) Full view HTML
Editorial
-
Article type: Editorial
2021Volume 18 Pages 289
Published: 2021
Released on J-STAGE: December 17, 2021
Advance online publication: December 02, 2021Download PDF (298K) Full view HTML
Regular Article
-
Article type: Regular Article
2021Volume 18 Pages 290-304
Published: 2021
Released on J-STAGE: December 18, 2021
Advance online publication: December 02, 2021 Download PDF (1876K) Full view HTML
Download PDF (1876K) Full view HTML -
Article type: Regular Article
2021Volume 18 Pages 305-316
Published: 2021
Released on J-STAGE: December 22, 2021
Advance online publication: December 04, 2021 Download PDF (967K) Full view HTML
Download PDF (967K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2021Volume 18 Pages 317-326
Published: 2021
Released on J-STAGE: January 08, 2022
Advance online publication: December 22, 2021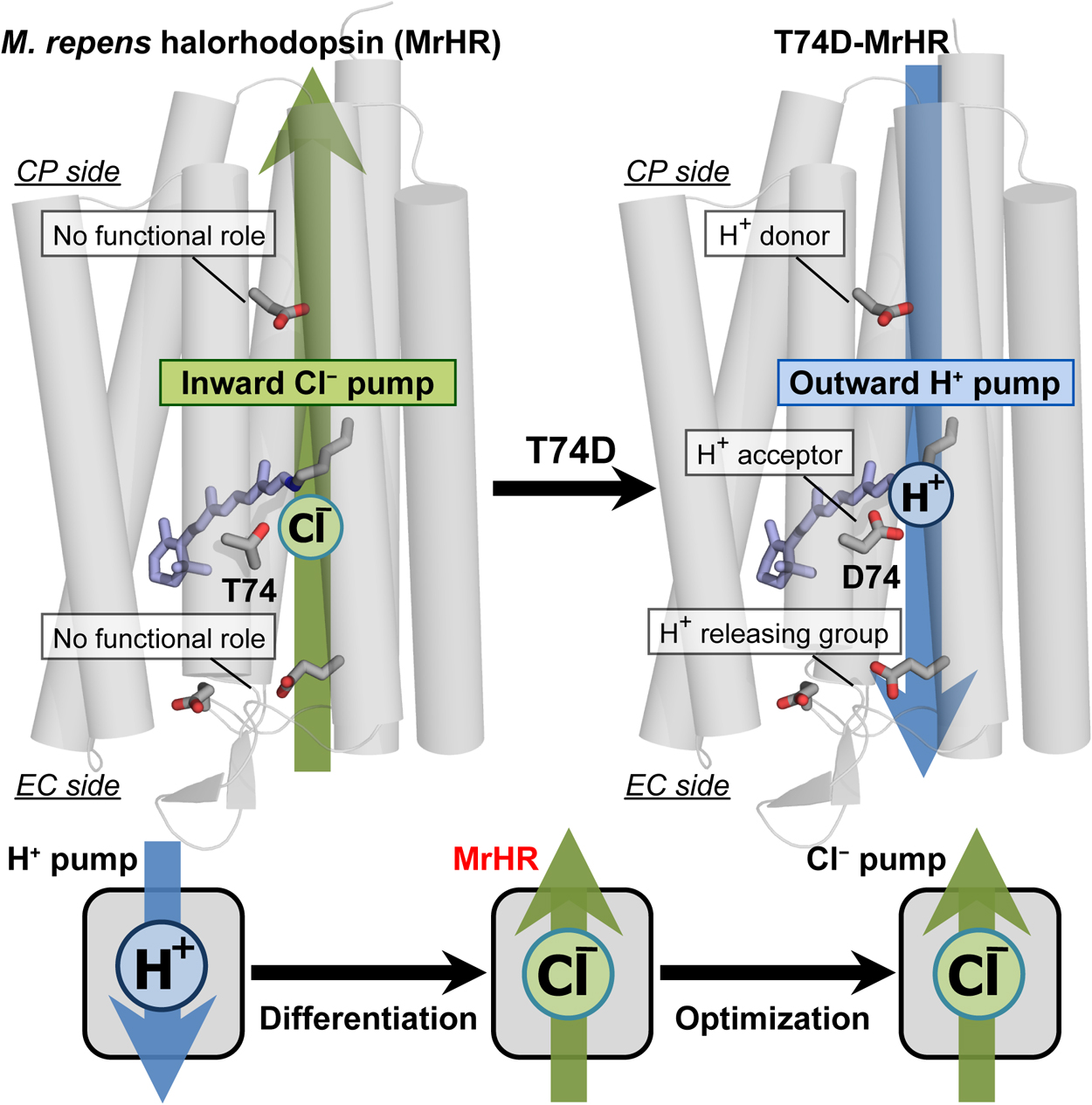 Download PDF (2683K) Full view HTML
Download PDF (2683K) Full view HTML
- |<
- <
- 1
- >
- >|