
Volume 18, Issue 4
Displaying 1-6 of 6 articles from this issue
- |<
- <
- 1
- >
- >|
Foreword
-
2019Volume 18Issue 4 Pages A13
Published: 2019
Released on J-STAGE: January 28, 2020
Download PDF (267K) Full view HTML
Highlight
-
2019Volume 18Issue 4 Pages A14-A20
Published: 2019
Released on J-STAGE: January 28, 2020
 Download PDF (4496K) Full view HTML
Download PDF (4496K) Full view HTML
General Paper
-
2019Volume 18Issue 4 Pages 169-175
Published: 2019
Released on J-STAGE: January 28, 2020
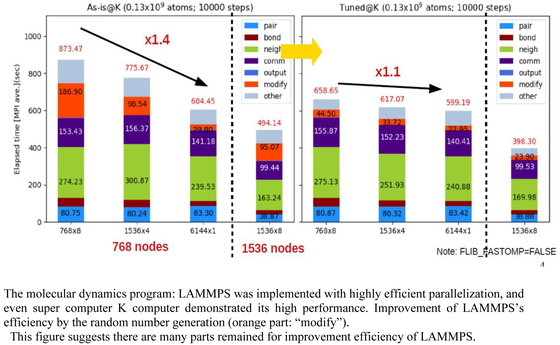 Download PDF (1383K) Full view HTML
Download PDF (1383K) Full view HTML -
2019Volume 18Issue 4 Pages 176-186
Published: 2019
Released on J-STAGE: January 28, 2020
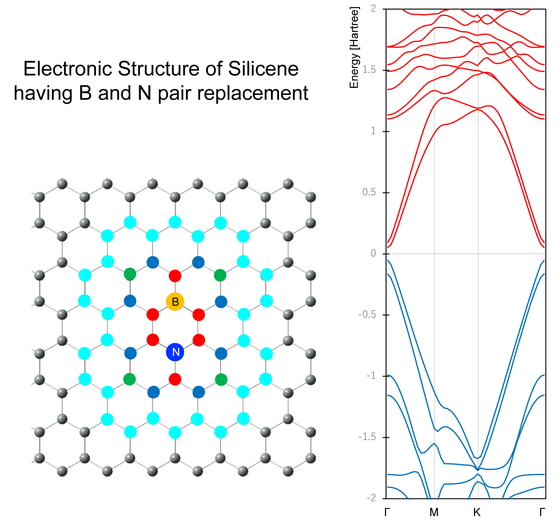 Download PDF (4492K) Full view HTML
Download PDF (4492K) Full view HTML -
2019Volume 18Issue 4 Pages 187-193
Published: 2019
Released on J-STAGE: January 28, 2020
Download PDF (1348K) Full view HTML -
2019Volume 18Issue 4 Pages 194-197
Published: 2019
Released on J-STAGE: February 07, 2020
Download PDF (509K) Full view HTML
- |<
- <
- 1
- >
- >|