
- |<
- <
- 1
- >
- >|
-
Masahiko HADA2021Volume 20Issue 1 Pages A1
Published: 2021
Released on J-STAGE: June 30, 2021
JOURNAL FREE ACCESS FULL-TEXT HTMLDownload PDF (362K) Full view HTML
-
Masahiko HADA2021Volume 20Issue 1 Pages A2-A10
Published: 2021
Released on J-STAGE: June 30, 2021
 JOURNAL FREE ACCESS FULL-TEXT HTML
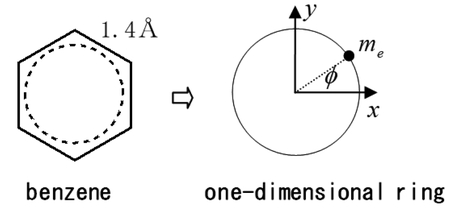
JOURNAL FREE ACCESS FULL-TEXT HTMLThe nucleus-independent chemical shift (NICS) of benzene is calculated using a one-dimensional ring model. In a real compound, an individual feature contributes to the NICS and it makes an intuitive understanding of the NICS complicate and difficult. We can avoid this complexity by using an extremely simple model. To overcome a classical and naive understanding such as the Fleming's left hand rule or the Ampère's circuital law, we apply the Schrödinger equation to an electron on a one-dimensional ring, and evaluate the NICS quantitatively in a framework of this ring model, using both Ramsey's equation and Pople's method. We also compare these results with the ab-initio MO calculations. From these results, we discuss about the diamagnetic and paramagnetic terms (diatropic- and paratropic-type currents) of NICS.
My Thoughts on the Education of Quantum Chemistry Fullsize ImageView full abstractDownload PDF (1087K) Full view HTML
-
Chiaki HANDA, Tomonaga OZAWA, Kaori FUKUZAWA, Etsuo YONEMOCHI2021Volume 20Issue 1 Pages 1-9
Published: 2021
Released on J-STAGE: June 30, 2021
 JOURNAL FREE ACCESS FULL-TEXT HTML
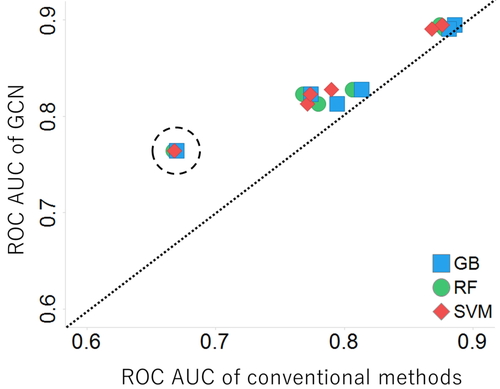
JOURNAL FREE ACCESS FULL-TEXT HTMLAmes test to detect mutagenicity in vitro is of crucial importance in drug discovery and development as an early alerting system for potential carcinogenicity and/or teratogenicity for drug candidates. In the alerting system of machine learning approaches, which are the main approach of in silico prediction, there is a concept called Applicability domain (AD) that has a significant influence on prediction accuracy. In drug discovery, prediction of drug candidate compounds with low structural similarity to the training data may take place, while such compounds have a high probability of being out of the AD and tend to be less accurate. In this study, we evaluated the performance of several machine learning methods for a group of compounds with a high probability of being included in AD or not, respectively. The results showed that the performance of the graph convolutional networks (GCN) method which have achieved a superior performance in a wide range of tasks was better than conventional methods. In particular, the accuracy of the GCN method is significantly different from the conventional methods for the prediction of molecules with low structural similarity (Figure 4).
View full abstractDownload PDF (1950K) Full view HTML -
Shunsuke ITO, Shigeru SAKURAZAWA2021Volume 20Issue 1 Pages 10-13
Published: 2021
Released on J-STAGE: July 17, 2021
JOURNAL FREE ACCESS FULL-TEXT HTMLAmino acid thermal heterocomplex molecules are primitive macromolecules synthesized by thermally polymerizing amino acids. Amino acid thermal heterocomplex molecules microspheres form capsules in response to changes in the surrounding environment, but the mechanism has not been clarified. In this study, we hypothesized that the autocatalytic properties of amino acid thermal heterocomplex molecules is a main factor in capsule formation and integrated a self- catalytic cluster formation mechanism into Brownian dynamics and tried to verify it. It was clarified that when cluster formation was incorporated, high-density regions were formed. This result suggests that the clusters in the high-density region grow further to form a capsule-like structure. From this result, amino acid thermal heterocomplex molecules, which is primitive macromolecules, has the function of the formation of physical compartment that is thought to have contributed to the origin of life, which is derived from the autocatalytic association process of the amino acid thermal heterocomplex molecules.
View full abstractDownload PDF (2117K) Full view HTML -
Wataru TAKAHARA, Yuki KOBAYASHI, Masashi MORITA, Kojiro OKUYAMA, Nobuy ...2021Volume 20Issue 1 Pages 14-21
Published: 2021
Released on J-STAGE: August 03, 2021
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLIn this study, we constructed machine learning models for predicting the relative permittivity (ε) and dielectric loss tangent (tanδ), which are important for the industrial application of thermosetting resin composites, using our own experimental data. We adopted a wide range of methods, including gradient boosting decision tree (GBDT) algorithms, which have been attracting attention in recent years, for the construction of machine learning models. Among the constructed models with multiple methods, we extracted models that satisfy the coefficient of determination R2CV > 0.8 at the time of cross-validation in the training data set. Furthermore, we selected the model in which the values of RMSE (Root Mean Square Error) and MAE (Mean Absolute Error) were small in the training data set and could predict the physical properties more quantitatively and evaluated them in the test data set. As a result, we obtained a machine learning model in which RMSE and MAE can predict physical properties on the order of 10−1 to 10−2 for the mean values of ε and tanδ, respectively. From this result, we have demonstrated for the first time that the approach by MI (Materials Informatics) is effective even for thermosetting resin composites and that quantitative property prediction is possible. We expect that the development period will be shortened and promoted using this developed MI approach.
View full abstractDownload PDF (2811K) Full view HTML
- |<
- <
- 1
- >
- >|