
Volume 16, Issue 5
Special Issue: Selected Papers from the Annual Autumn Meeting 2017
Displaying 1-20 of 20 articles from this issue
- |<
- <
- 1
- >
- >|
Foreword
-
2017 Volume 16 Issue 5 Pages A29-A30
Published: 2017
Released on J-STAGE: January 30, 2018
Download PDF (311K) Full view HTML
Letters (Selected Paper)
-
2017 Volume 16 Issue 5 Pages 119-122
Published: 2017
Released on J-STAGE: January 30, 2018
 Download PDF (1089K) Full view HTML
Download PDF (1089K) Full view HTML -
2017 Volume 16 Issue 5 Pages 123-125
Published: 2017
Released on J-STAGE: January 30, 2018
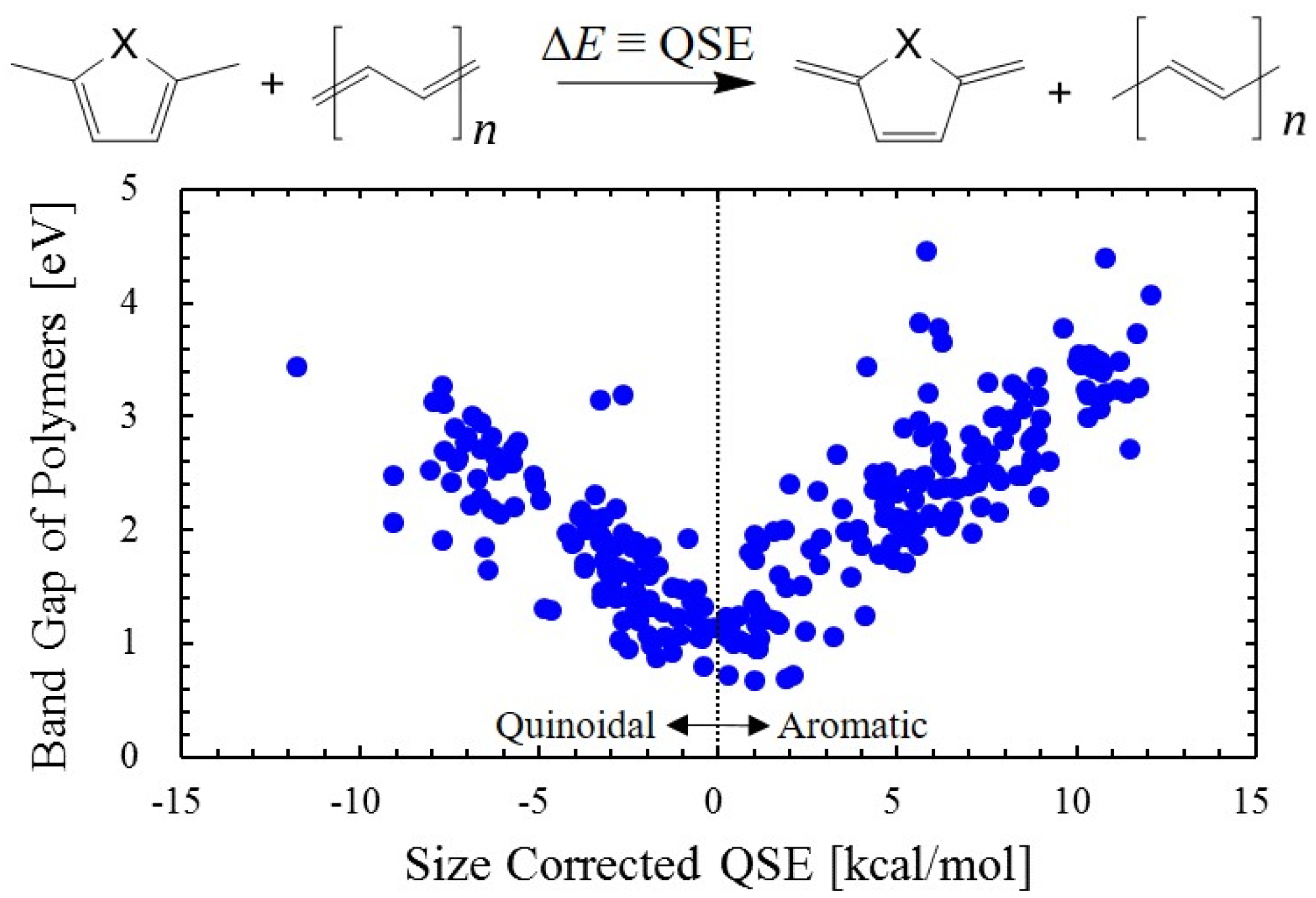 Download PDF (1041K) Full view HTML
Download PDF (1041K) Full view HTML -
2017 Volume 16 Issue 5 Pages 126-128
Published: 2017
Released on J-STAGE: February 10, 2018
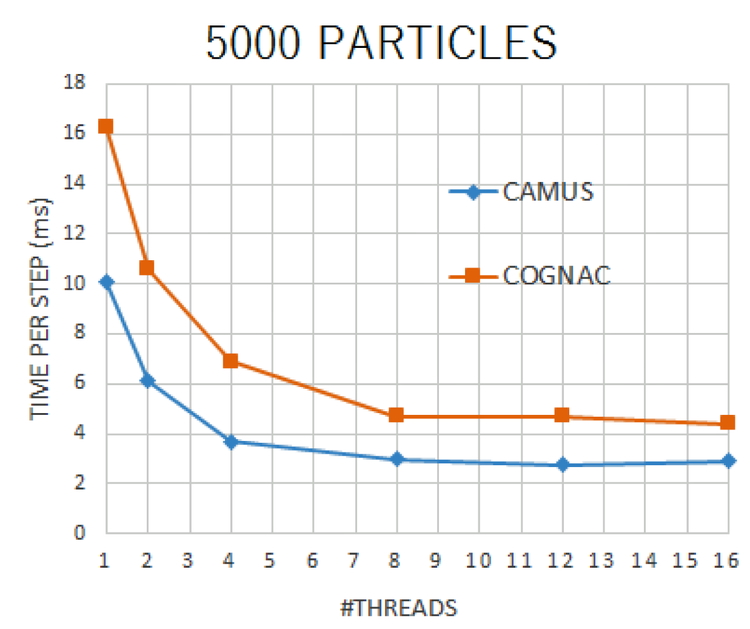 Download PDF (438K) Full view HTML
Download PDF (438K) Full view HTML -
2017 Volume 16 Issue 5 Pages 129-130
Published: 2017
Released on J-STAGE: February 12, 2018
Download PDF (1426K) Full view HTML -
2017 Volume 16 Issue 5 Pages 131-132
Published: 2017
Released on J-STAGE: February 10, 2018
 Download PDF (1565K) Full view HTML
Download PDF (1565K) Full view HTML -
2017 Volume 16 Issue 5 Pages 133-134
Published: 2017
Released on J-STAGE: February 10, 2018
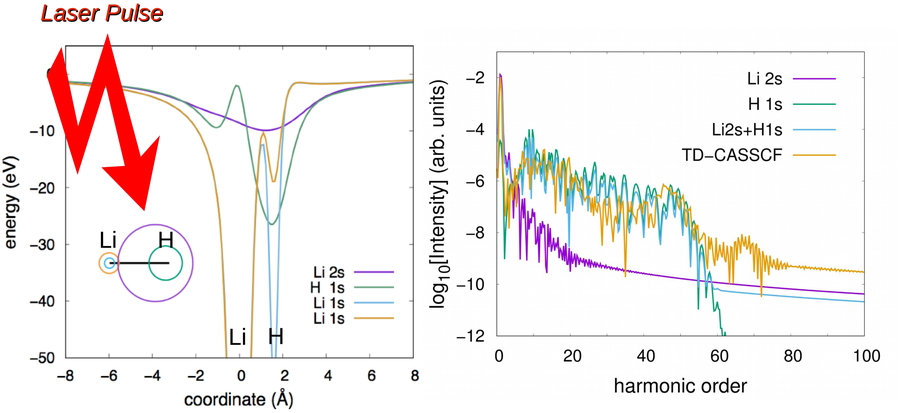 Download PDF (770K) Full view HTML
Download PDF (770K) Full view HTML -
2017 Volume 16 Issue 5 Pages 135-138
Published: 2017
Released on J-STAGE: February 20, 2018
 Download PDF (986K) Full view HTML
Download PDF (986K) Full view HTML -
2017 Volume 16 Issue 5 Pages 139-140
Published: 2017
Released on J-STAGE: February 20, 2018
 Download PDF (434K) Full view HTML
Download PDF (434K) Full view HTML -
2017 Volume 16 Issue 5 Pages 141-143
Published: 2017
Released on J-STAGE: February 20, 2018
 Download PDF (831K) Full view HTML
Download PDF (831K) Full view HTML -
2017 Volume 16 Issue 5 Pages 144-146
Published: 2017
Released on J-STAGE: February 20, 2018
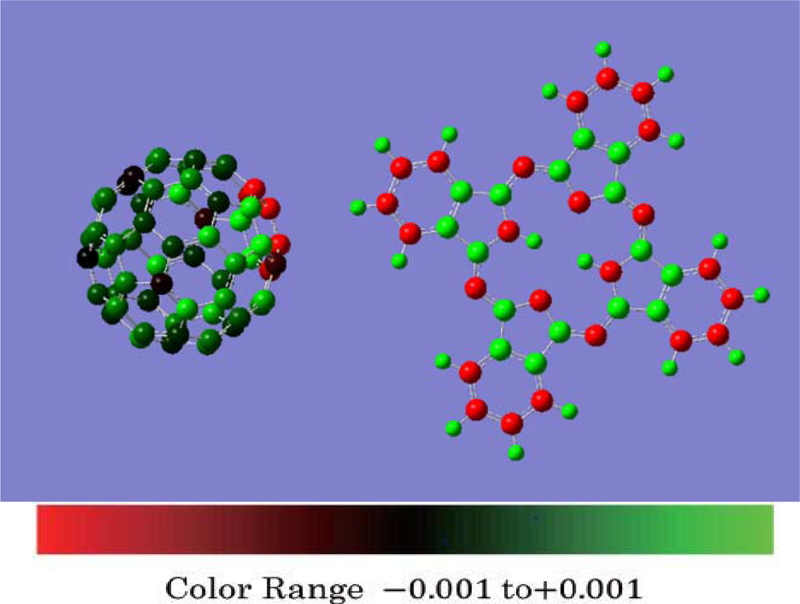 Download PDF (890K) Full view HTML
Download PDF (890K) Full view HTML -
2017 Volume 16 Issue 5 Pages 147-148
Published: 2017
Released on J-STAGE: February 24, 2018
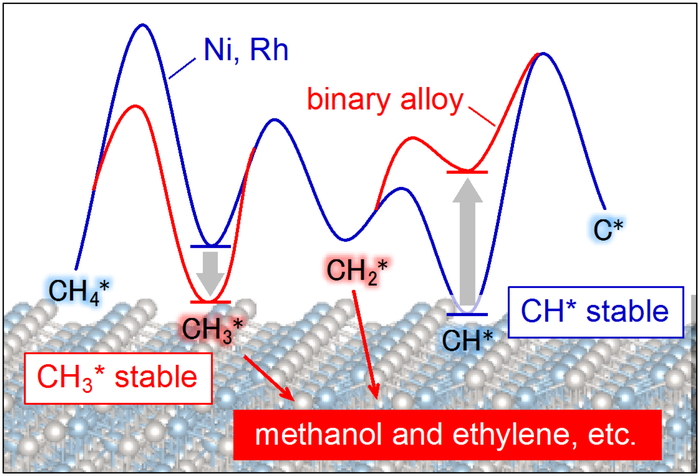 Download PDF (507K) Full view HTML
Download PDF (507K) Full view HTML -
2017 Volume 16 Issue 5 Pages 149-151
Published: 2017
Released on J-STAGE: February 24, 2018
 Download PDF (1730K) Full view HTML
Download PDF (1730K) Full view HTML -
2017 Volume 16 Issue 5 Pages 152-154
Published: 2017
Released on J-STAGE: March 07, 2018
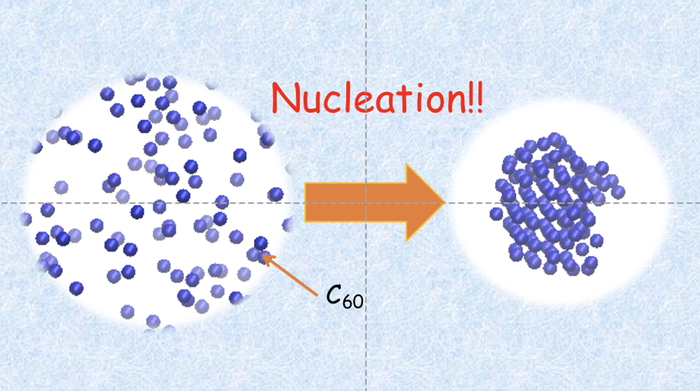 Download PDF (848K) Full view HTML
Download PDF (848K) Full view HTML -
2017 Volume 16 Issue 5 Pages 155-156
Published: 2017
Released on J-STAGE: March 07, 2018
 Download PDF (427K) Full view HTML
Download PDF (427K) Full view HTML -
2017 Volume 16 Issue 5 Pages 157-159
Published: 2017
Released on J-STAGE: March 13, 2018
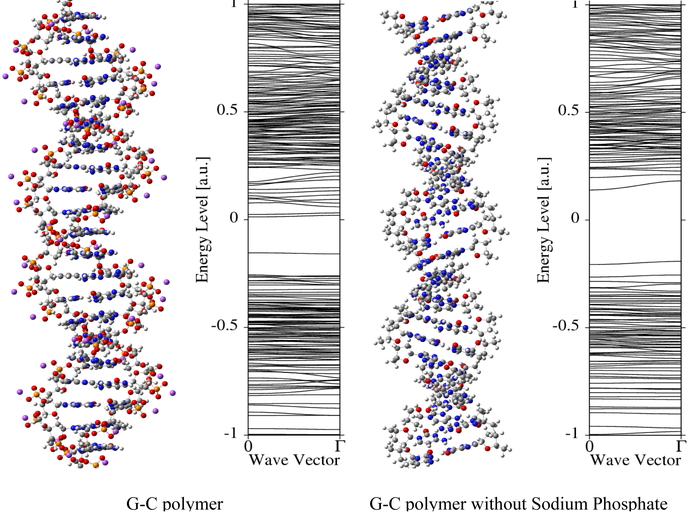 Download PDF (842K) Full view HTML
Download PDF (842K) Full view HTML -
2017 Volume 16 Issue 5 Pages 160-162
Published: 2017
Released on J-STAGE: April 06, 2018
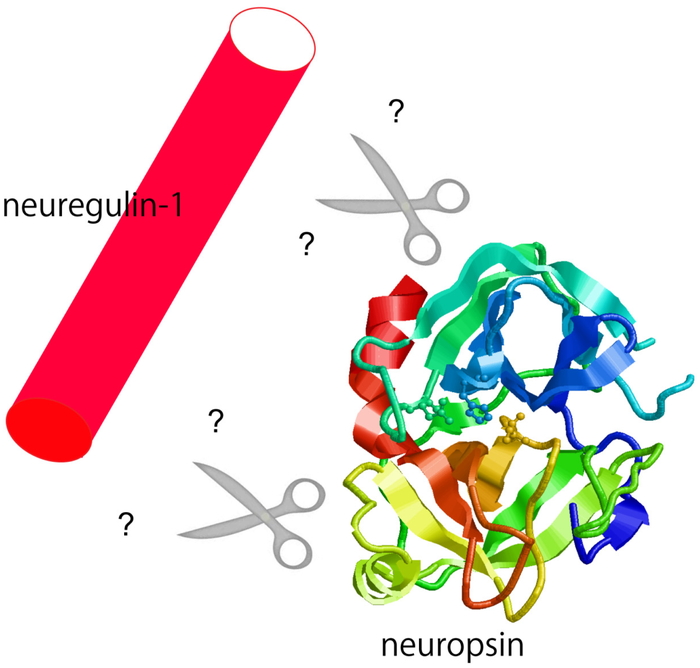 Download PDF (1271K) Full view HTML
Download PDF (1271K) Full view HTML -
2017 Volume 16 Issue 5 Pages 163-164
Published: 2017
Released on J-STAGE: April 06, 2018
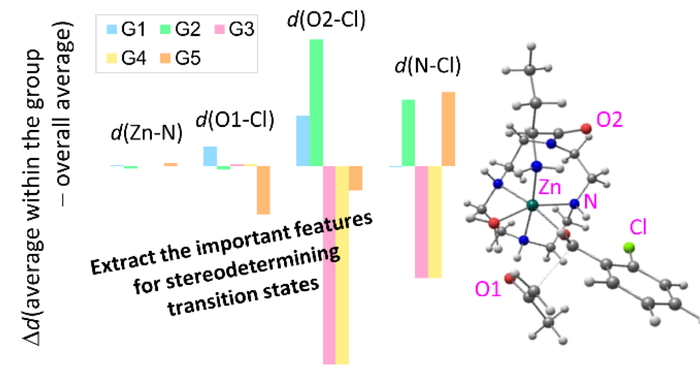 Download PDF (618K) Full view HTML
Download PDF (618K) Full view HTML -
2017 Volume 16 Issue 5 Pages 165-166
Published: 2017
Released on J-STAGE: April 10, 2018
 Download PDF (731K) Full view HTML
Download PDF (731K) Full view HTML -
2017 Volume 16 Issue 5 Pages 167-169
Published: 2017
Released on J-STAGE: April 18, 2018
Download PDF (502K) Full view HTML
- |<
- <
- 1
- >
- >|