
Volume 17, Issue 3
Special Issue: Selected Papers from the Annual Spring Meeting 2018
Displaying 1-19 of 19 articles from this issue
- |<
- <
- 1
- >
- >|
Foreword
-
2018Volume 17Issue 3 Pages A15
Published: 2018
Released on J-STAGE: October 06, 2018
Download PDF (316K) Full view HTML
Letters (Selected Paper)
-
2018Volume 17Issue 3 Pages 111-112
Published: 2018
Released on J-STAGE: October 06, 2018
 Download PDF (1863K) Full view HTML
Download PDF (1863K) Full view HTML -
2018Volume 17Issue 3 Pages 113-116
Published: 2018
Released on J-STAGE: October 06, 2018
 Download PDF (933K) Full view HTML
Download PDF (933K) Full view HTML -
2018Volume 17Issue 3 Pages 117-119
Published: 2018
Released on J-STAGE: October 10, 2018
 Download PDF (768K) Full view HTML
Download PDF (768K) Full view HTML -
2018Volume 17Issue 3 Pages 120-121
Published: 2018
Released on J-STAGE: October 16, 2018
Download PDF (497K) Full view HTML -
2018Volume 17Issue 3 Pages 122-123
Published: 2018
Released on J-STAGE: October 16, 2018
 Download PDF (573K) Full view HTML
Download PDF (573K) Full view HTML -
2018Volume 17Issue 3 Pages 124-126
Published: 2018
Released on J-STAGE: October 16, 2018
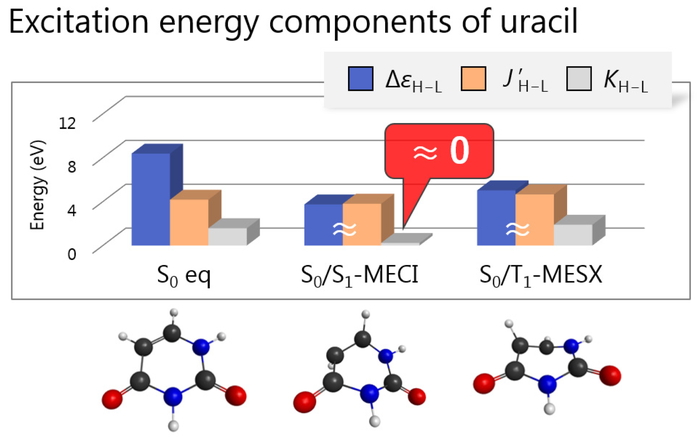 Download PDF (1108K) Full view HTML
Download PDF (1108K) Full view HTML -
2018Volume 17Issue 3 Pages 127-129
Published: 2018
Released on J-STAGE: October 16, 2018
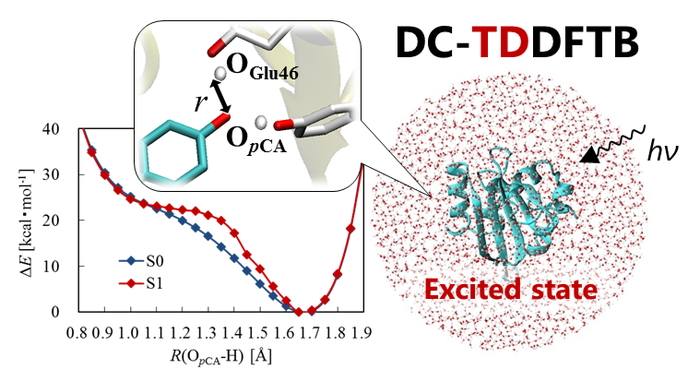 Download PDF (964K) Full view HTML
Download PDF (964K) Full view HTML -
2018Volume 17Issue 3 Pages 130-132
Published: 2018
Released on J-STAGE: October 16, 2018
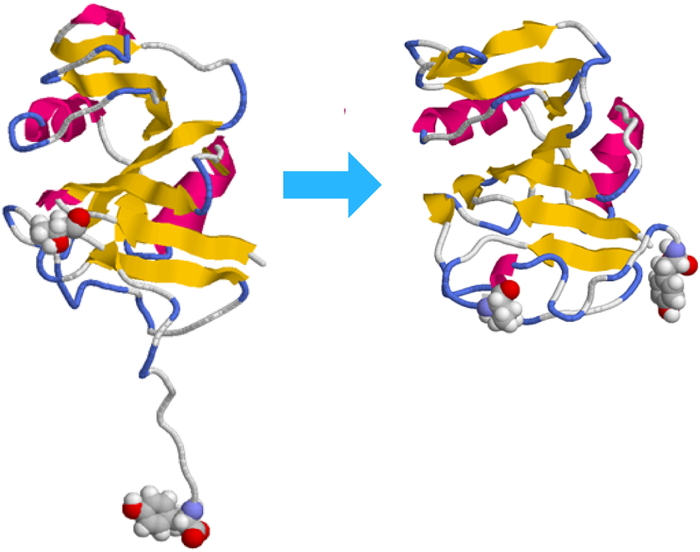 Download PDF (960K) Full view HTML
Download PDF (960K) Full view HTML -
2018Volume 17Issue 3 Pages 133-137
Published: 2018
Released on J-STAGE: October 16, 2018
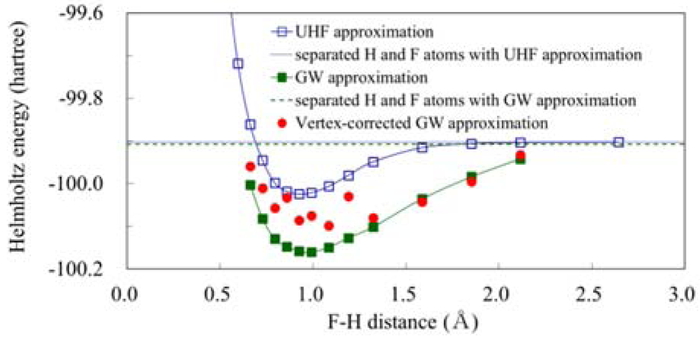 Download PDF (740K) Full view HTML
Download PDF (740K) Full view HTML -
2018Volume 17Issue 3 Pages 138-141
Published: 2018
Released on J-STAGE: October 22, 2018
 Download PDF (1573K) Full view HTML
Download PDF (1573K) Full view HTML -
2018Volume 17Issue 3 Pages 142-143
Published: 2018
Released on J-STAGE: October 26, 2018
 Download PDF (495K) Full view HTML
Download PDF (495K) Full view HTML -
2018Volume 17Issue 3 Pages 144-146
Published: 2018
Released on J-STAGE: October 26, 2018
 Download PDF (1372K) Full view HTML
Download PDF (1372K) Full view HTML -
2018Volume 17Issue 3 Pages 147-149
Published: 2018
Released on J-STAGE: November 02, 2018
 Download PDF (1037K) Full view HTML
Download PDF (1037K) Full view HTML -
2018Volume 17Issue 3 Pages 150-152
Published: 2018
Released on J-STAGE: November 10, 2018
 Download PDF (561K) Full view HTML
Download PDF (561K) Full view HTML -
2018Volume 17Issue 3 Pages 153-154
Published: 2018
Released on J-STAGE: November 10, 2018
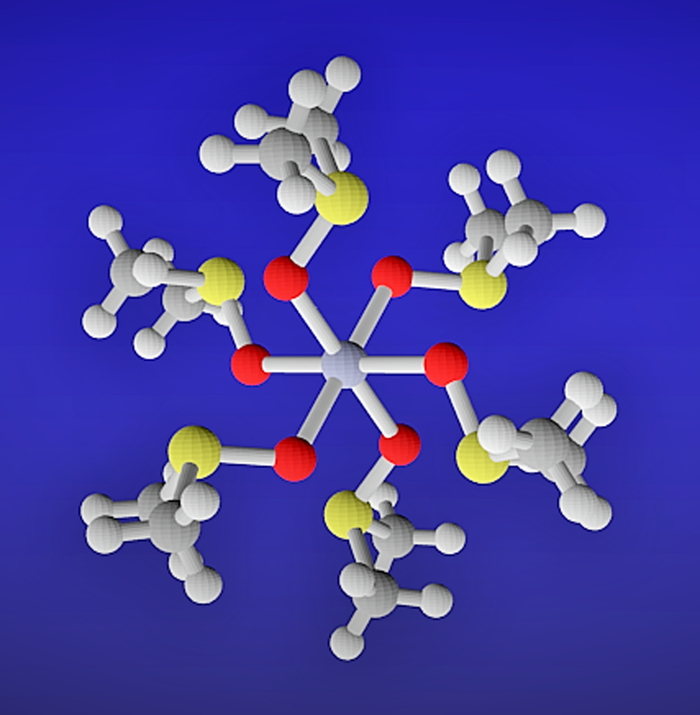 Download PDF (689K) Full view HTML
Download PDF (689K) Full view HTML -
2018Volume 17Issue 3 Pages 155-157
Published: 2018
Released on J-STAGE: November 10, 2018
 Download PDF (1282K) Full view HTML
Download PDF (1282K) Full view HTML -
2018Volume 17Issue 3 Pages 158-159
Published: 2018
Released on J-STAGE: November 17, 2018
Download PDF (1523K) Full view HTML -
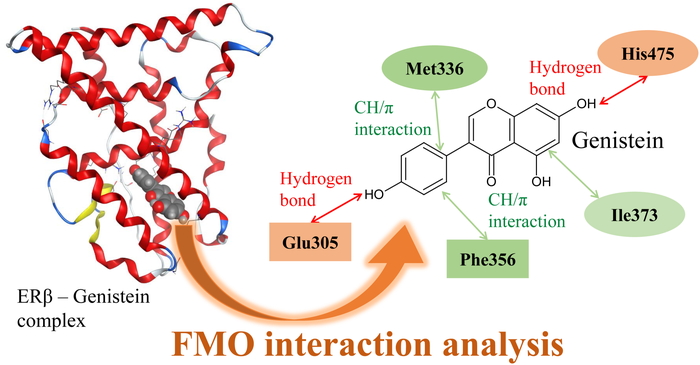
Analysis of ligand binding specificity of estrogen receptor by the fragment molecular orbital method2018Volume 17Issue 3 Pages 160-162
Published: 2018
Released on J-STAGE: December 21, 2018
 Download PDF (1044K) Full view HTML
Download PDF (1044K) Full view HTML
- |<
- <
- 1
- >
- >|