-
大石 正道, 大坂 真起子, 佐藤 守, 小寺 義男, 前田 忠計, 酒井 康弘, 古舘 専一
セッションID: 1G1-1
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
The
rdw rat has hereditary hypothyroidism caused by a missense mutation (G2320R) with thyroglobulin (Tg) gene account for the Tg misfolding. In this report we revealed proteomic changes in the
rdw adrenal gland by using an agarose two-dimensional gel electrophoresis (2-DE) and a liquid chromatography-tandem mass spectrometry system (LC-MS/MS). We compared the proteome map of the
rdw adrenal cortex and medulla with that of normal tissues, quantitative alteration of protein contents related to adrenal function were detected, and the abnormal proteins were identified. In the
rdw adrenal cortex, at least 7 protein spot levels were decreased. One of those identified proteins was 11beta-hydroxysteroid dehydrogenase (11beta-HSD) known to be the enzyme converting 11-deoxycorticosterone to corticosterone. In the
rdw adrenal medulla, at least 2 protein spot levels were significantly increased. These proteins were respectively identified as tyrosine 3-hydroxylase (TH), which catalyzes the rate-limiting step in catecholamine biosynthesis, and secretogranin II (SgII), known as a neuroendocrine secretory granule protein. All the above alterations of protein contents were recovered by administration of T
4 to
rdw rat from three days to 28 days of age, however, resulted in failure by normal thyroid transplantation at 28 days of age. These results suggest that neonatal thyroid status critically affects expression of those proteins related to adrenal function.
抄録全体を表示
-
朝長 毅, 松下 一之, 大石 正道, 小寺 義男, 前田 忠計, 島田 英昭, 落合 武徳, 野村 文夫
セッションID: 1G1-2
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Purpose: Although numerous proteome studies have recently been performed to identify cancer-related changes in protein expression, only a limited display of relatively abundant proteins has been identified. The aim of this study is to identify novel proteins as potential tumor markers in primary colorectal cancer tissues by a high-resolution two-dimensional gel electrophoresis (2-DE).
Experimental Design: 2-DE using an agarose gel for isoelectric focusing was used to compare protein profiling of ten colorectal cancer tissues and adjacent normal mucosa. Altered expression and post-translational modification of several proteins was examined by Western blotting and immunohistochemistry.
Results: 97 proteins out of 107 spots (90.7%) which were differentially expressed between matched normal and tumor tissues were identified by mass spectrometry. Among them, 42 unique proteins (49spots) significantly increased or decreased in the tumors. They include eukaryotic translation initiation factor 4H (eIF-4H), inorganic pyrophosphatase, anterior gradient 2 homologue (hAG-2), aldolase A and chloride intracellular channel 1 (NCC27), whose elevated expression in tumor tissues was confirmed by Western blotting and immunohistochemistry. Interestingly, only isoform 1 of two transcript variants of eIF-4H was greatly upregulated in most of the tumor tissues. Moreover, post-translational modifications of the prolyl-4-hydroxylase b subunit (P4HB) and annexin A2 were also identified.
Conclusions: We identified several novel proteins with altered expression in primary colorectal cancer using agarose 2-DE. This method is a powerful technique with which to search for not only quantitative but also qualitative changes in a biological process of interest and may contribute to the deeper understanding of underlying mechanisms of human cancer.
抄録全体を表示
-
中村 和行, 藤本 正憲, 蔵満 保宏, 長坂 祐二, 山崎 雄三, 栗木 智子, 戸田 年総, ソ-ベル アンドレ
セッションID: 1G1-3
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Two-dimensional gel electrophoresis (2DE) and matrix-assisted laser desorption/ionization-mass spectrometry (MALDI-MS) were used for proteome analysis on heat stress-induced apoptosis in Jurakat cells of a human T lymphoblastic leukemia cell line. The heat-stress was given by incubating cells for 30 min at 43 C up to 49 C in a serum free culture medium. Intracellular soluble proteins were extracted from the cells immediately after the incubation and submitted to 2DE. Stathmin phosphorylated forms, calmodulin, protein kinase C substrate and thymosin beta-4 were increased in temperature dependent manner although heat shok protein 70 was not increased by the incubation. On the other hand, ubiquitin-like protein SMT3B, eukaryote translation initiation factor-5A and -3 subunit, Rho GDP-dissociation inhibitor 1, and protein phosphatase 2C were significantly decreased. Further examinations by MALDI/MS/MS more showed that the temperature dependent phosphorylation of stathmin was initially occured at Ser37 specific site for CDK-1 and followed by phosphorylation at Ser24 for CDK-1/MAPK or Ser15 for PKA at higher temperature.
抄録全体を表示
-
請川 亮, 平野 久, 鮎沢 大
セッションID: 1G1-4
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
5-Bromdeoxyuridine (BrdU) immediately and clearly induces a phenomenon similar to premature senescence in any type of mammalian cells. To understand cellular senescence, characterization of genes is not sufficient because protein levels markedly fluctuate during cellular senescence and ageing. Since BrdU seems to target chromatin structure to affect expression of senescence-associated genes, we made an attempt to characterize nuclear proteins in HeLa cells undergoing senescence with BrdU by 2D DIGE.
We purified nuclei from HeLa cells cultured with BrdU for 4 days and control cells and prepared samples of soluble nuclear proteins. These samples were subjected to DIGE and the data were processed to find protein spots altered upon addition of BrdU. Sevral spots were found to alter reproducibly. Of such spots, lamin A and C were markedly increased and members of the hnRNA family markedly decreased. Levels of mRNA for these genes did not significantly change upon addition of BrdU. The changes in the lamin proteins seem to agree with the observation that nuclei enormously swell during cellular senescence. Recently, it is also shown that Hutchinson-Gilford progeria syndrome is caused by a mutation in the LMNA gene encoding lamin A/C. An abnormal lamin A protein is found to lead to nuclear swelling and a defect of nuclear lamina in cells from patients. We thus performed Western blot analysis to characterize lamin A/C in Hela cells using specific antibody. Interestingly, an abnormal lamin A protein was found to accumulate in BrdU-treated cells. The hnRNA family proteins are involved in various functions. We found that a member of the hnRNP family is responsible for stress response that leads to senescence.
Based on close similarity between BrdU-mediated senescence in immortal cells and replicative senescence in normal human cells, it is suggested that lamin A/C and hnRNPs play a central role in cellular senescence.
抄録全体を表示
-
平林 淳
セッションID: 1G2-1
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
In order to elucidate precise functions of glycans attached to extensive proteins, it is necessary to develop a fundamental database of glycoproteins, which is annotated as regards not only genes that encode core proteins and glycosylation sites, but also glycan structures. Various techniques have now been attempted to achieve this with the aid of advanced procedures of mass spectrometry. However, it has not yet been achieved in a fully satisfactory manner. In the course of the NEDO project "Structural Glycomics" (2002.3-2005.3), we have started developing a total system for glycoproteomics by collaboration of GL Sciences on the basis of a recently established procedure named "glyco-catch", which enables selective capture of a set of glycopeptides by lectin columns followed by a large scale of identification of glycoprotein genes and Asn-linked glycosylation sites by an improved proteomic MS/MS procedure (1,2). To further profile glycan structures, however, the above strategy must be much improved as regards in particular 1) manipulation of a number of glycopeptides separated by a 2D-LC system, each of which is subjected to peptide-N-glycanse treatment to liberate glycans, and 2) selective recovery of peptides and glycans for further structural analysis by either MS or lectin profiling. In the session, the core strategy is described with a few experiments using a model glycoprotein and actual samples of mouse glycoproteins captured by a ConA-column.
(1) Hirabayashi, J., Kaji, H., Isobe, T., and Kasai K. J. Biochem (Tokyo) 132, 103 - 114, 2002
(2) Kaji, H., Saito, H., Yamauchi, Y., Shinkawa, T., Taoka, M., Hirabayashi, J., Kasai, K., Takahashi, N., Isobe, T. Nat Biotechnol. 21, 667-672, 2003
抄録全体を表示
-
川崎 ナナ, 原園 景, 橋井 則貴, 伊藤 さつき, 川西 徹, 早川 堯夫
セッションID: 1G2-2
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Oligosaccharides play important roles in diverse biological processes, and alterations in them are thought to be associated with some diseases. The elucidation of glycosylation-related diseases will require glycomic approaches based on qualitative and quantitative analyses of glycosylation. We have studied the various analytical methods to determine carbohydrate heterogeneity, glycosylation sites, and site-specific glycosylation in glycoproteins by using LC/MS.
LC/MS/MS of glycopeptidesMass spectrometric peptide/glycopeptide mapping is frequently used for the determination of glycosylation sites and site-specific glycosylation. In-source fragmentation and precursor ion scanning, which monitor sugar-specific oxonium ions, are employed to extract glycopeptide ions from a large number of peptide ions, however, additional analyses are required in order to obtain peptide fragmentation patterns or precursor ions. Using automated data-dependent MS/MS as an alternative method to distinguish glycopeptide ions from peptide ions and to obtain both the peptide's and sugar's fragment ions, we have successfully determined the site-specific glycosylation of some glycoproteins.
LC/MS of oligosaccharidesGlycoproteins exist in heterogeneous forms because of the various combinations of oligosaccharides including isomers. We have developed mass spectrometric oligosaccharide profiling using a graphitized carbon column, which can separate oligosaccharides based on subtle differences in branch, position, and linkage isomers with volatile solution. LC/MS provides structural information based on chromatographic behavior and molecular mass. This method is expected to be a powerful tool for glycome analysis. In this study we apply oligosaccharide profiling to differential analyses of N-linked oligosaccharides between treated and untreated cells.
抄録全体を表示
-
掛樋 一晃
セッションID: 1G2-3
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
We proposed an approach to categorize carbohydrate chains in glycoconjugates using capillary affinity electrophoresis (CAE). CAE allows the determination of the interaction between carbohydrate chains and proteins (Nakajima et al. J. Proteome Res. 2003), and specific affinity of an oligosaccharide toward a lectin results resolution of complex mixture of oligosaccharides. In the present lecture, the principle of capillary affinity electrophoresis is briefly shown for categorizing carbohydrate chains or detection of carbohydrate-binding proteins in biological samples. And some applications are also shown for detection of novel carbohydrates in serum samples of cancer patients. An example to detect a carbohydrate-binding protein is also shown. 1. We can simultaneously determine the carbohydrate chains of high-mannose, complex and hybrid type oligosaccharides in biological samples using some defined sets of lectins (ConA, TGA, DSA, LTA, UEA and RSL) with extremely high sensitivity. Total time required for the analysis of an oligosaccharide mixture derived from one glycoprotein sample was within 2 h. Total amount required for the analysis was 200 ng (5 pmol) as glycoprotein. It should be noticed that the present method does not need purification of each oligosaccharide prior to CAE.2. We found marked changes in carbohydrate chains of alpha1-acid glycoprotein (AGP) in sera under malignancy. Alternations in branching patterns of carbohydrate chains and appearance of hyperfucosylated carbohydrate chains were especially interesting. These changes of carbohydrate chains in AGP may reflect the physiological environment in malignancy.
抄録全体を表示
-
和田 芳直, 田尻 道子
セッションID: 1G2-4
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Strategy for characterizing glycoforms and their attachment sites of glycoprotein (Summary)
I.
Finding glycopeptides in the peptide/glycopeptide mixtures(a) Glycan detection with oxonium ions in LC-MS: In-source fragmentation at the ESI source generates the glycan-specific oxonium ions such as HexNAc (m/z204), HexNAc-Hex (m/z366), HexNAc-Hex-NANA (m/z657). The selected ion monitoring for these ions allows to detect glycopeptides in the LC eluents. However, duplicate run is necessary to execute on-line LC-MS/MS. Otherwise, the corresponding fractions are collected for off-line analyses.
(b) MS of the glycopeptides isolated by lectin affinity chromatography: Concanavalin A-agarose is used to collect glycopeptides. The bound glycopeptides are eluted with mannose and subjected to MALDI- or ESI-MS. This is a facile way to analyze glycopeptides, while the universal binding of all glycan forms by single lectin is unavailable.
(c) Mixture analysis of glycoprotein digests: ESI- or MALDI-MS of digest is applicable to small glycoproteins. However, the availability become limited as the glycoprotein or the derived glycopeptide is larger in size.
II.
Determining both glycoforms and attachment sitesThe multiple tandem MS named MS
n is a technique enabling the glycan and peptide analyses in a single mass spectrometer. MALDI-QIT-TOF-MS generates a series of product ions for "ladder" sequence of glycoforms by the dissociation of the glycan moiety of a precursor glycopeptide. The ions of bare peptide backbone in the mass spectrum is then selected as a precursor for dissociation to deduce amino acid sequence. This technique is sensitive as to require only a few micrograms of glycoprotein as the starting sample.
抄録全体を表示
-
山下 亮, 藤原 優子, 安田 和基, 鏑木 康志
セッションID: 1G3-1
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
It is known that adipocytes secrete proteins called adipocytokines, which take part in the regulation of the lipid and glucose metabolism. Similar to adipocytes tissue, liver is recently speculated to secrete proteins, which have some physiological functions. The hepatic lipid homeostasis is regulated by a cross talk between peroxisome proliferator-activated receptor (PPAR) alpha and liver X receptor (LXR), both of which are nuclear receptors. We attempted to identify the secreted proteins, which are regulated by PPAR-alpha or LXR in hepatocytes using proteomic analysis. Human hepatoma HepG2 cells were treated with TO-901317 (LXR agonist) or Wy-14, 643 (PPAR-alpha agonist). After 36-48 h, media were collected and concentrated by centrifugal filter devices. Paired samples from cells treated with vehicle or ligands were labeled with fluorescent Cy3 or Cy5 dyes and electrophoresed on the same two-dimensional gels. Some spots from the ligand-treated sample were found to be upregulated or downregulated as compared with those from vehicle sample. For identification of these secreted proteins by mass spectrometry, the 2-DE gel, loaded with 0.5-1 mg of total protein, was stained with Sypro Ruby. After in-gel digestion, the peptides from the protein spots were analyzed using RPLC-ESI MS/MS. We found that one of the proteins with significant differences was identified as apolipoproteinE, secretion of which was previously shown to be increased by treatment with LXR agonist. Now, we are trying to identify other secreted proteins from hepatocytes, which may have some roles in regulating lipid metabolism.
抄録全体を表示
-
藏滿 保宏, 高島 元成, 横山 雄一郎, 飯塚 徳男, 戸田 年総, 坂井田 功, 沖田 極, 岡 正朗, 中村 和行
セッションID: 1G3-2
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Hepatocellular carcinoma (HCC) is one of the most common fatal cancers worldwide. The most clearly established risk factor for HCC is chronic infection with hepatitis B or C virus (HBV or HCV). The characteristic of HCC in Japan is that they originate from the liver tissues of hepatitis HCV or liver cirrhosis. Therefore, it is important to elucidate the factors related to the products from tumor tissues of HCV-infected patients, and to identify some proteins with which we can speculate the mechanism of carcinogenesis of HCC. Liver tissue samples of HCC cancerous tissues or non-cancerous tissues from patients infected with HCV were prepared and separated by means of two-dimensional gel electrophoresis. From proteomic differential display analysis, 11 spots whose expression in cancerous tissues increased, and 11 spots whose expression in tumor tissues decreased, were detected. Eight proteins out of 11 decreasing spots were identified as liver type aldolase, tropomyosin b-chain, ketohexokinase, enoyl-CoA hydratase, albumin, smoothelin, ferritin light chain, and arginase 1. Nine proteins out of 11 increasing spots were identified as GRP78, HSC70, GRP75, HSP70.1, HSP60, ATP synthase b-chain, glutamine synthetase(GS), phosphoglycerate mutase 1, and triosephosphate isomerase. The three protein spots of the same molecular weight (42kDa), whose expression increased in well-differentiated cancerous tissues, were identified as GS. The tryptic peptides of the most acidic GS isoform lost the signal of 899.5 Da corresponding a peptide of SASIRIPR and gained the signal of 1059.5 Da which was submitted to post source decay (PSD) analysis. The PSD analysis suggested the neutral loss by elimination of two phosphate groups which were supposed to be on serine residues of 899.5 Da peptide from serine 320 to arginine 327 in GS. The PMF followed by PSD analysis is thought to be useful for the determination of phosphorylation site of proteins showing molecular heterogeneity.
抄録全体を表示
-
鍋谷 卓司, 田伏 洋, 次田 晧, 正田 純一
セッションID: 1G3-3
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Primary intrahepatic calculosis (IHC), which is characterized by the formation of stones in the hepatic bile duct, is an intractable liver disease and suspected to be one of the causes of cholangiocellular carcinoma. To obtain an insight into the disease, we performed proteomic analysis of the normal and the affected hepatic tissues from patients with primary intrahepatic calculosis by two-dimensional electrophoresis followed by peptide mass fingerprinting. For the normal tissue, 195 spots out of 613 spots were identified as proteins, defining unique 137 proteins. For the IHC tissue, 137 spots out of 671 spots were identified as proteins, defining unique 101 proteins. To further precisely compare, we used two-dimensional difference gel electrophoresis (2-D DIGE). Consequently we identified the up-regulated proteins that were 12 and the down-regulated proteins that were 22. The up-regulated proteins contained the proteins related to plasma and inflammation. The down-regulated proteins contained 60 kDa heat shock protein, annexin VI, glutathione S-transferase A2.
抄録全体を表示
-
辻 輝之, 塩崎 愛子, 青木 雅弘, 下濱 俊
セッションID: 1G4-1
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Quantitative proteome analysis was performed in Alzheimer disease brain with 2-D gel to identify disease specific changes in protein expression. These changes were also investigated in serum and urine of Alzheimer patients. The task of characterizing the proteome and its components is now practically achievable because of the development and integration of four important tools; complete genome sequence databases, mass spectrometry, matching software for protein sequences and protein separation technology. Mass spectrometry instrumentation has undergone tremendous change over the past decade, culminating in the development of highly sensitive, robust instruments that can reliably analyze biomolecules, particularly proteins and peptides. In brain protein separation, we sequentially extracted them using two distinct sample solutions, yielding different protein fractions. These fractions showed distinct 2-DE patterns with high resolution and excellent reproducibility. In soluble fraction, approximately 1300 protein spots were detected, and five spots are significantly increased and 10 spots are significantly decreased in AD. The proteins identified include enzymes, molecular chaperones and cytoskeletal proteins. In low soluble fraction, over 500 protein spots were detected in the 2-DE data analysis. There were three spots significantly increased in AD. Two of these spots were identified as glial fibrillary acidic protein using mass spectrometry. These findings prompt further study on disease-linked proteins for the investigation of AD pathogenesis and for the quest of disease markers in serum and urine.
抄録全体を表示
-
戸田 年総, 荒木 令江, 久富 寿, 川野 克己, 森澤 拓, 廣田 美佳子
セッションID: 1G4-2
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Protein phosphorylation is a typical posttranslational modification of protein, which plays important role in various metabolic regulation and signal transduction. Indeed, abnormal phosphorylation of key control protein has been detected in many cascades and is thought to underlie the associated cellular dysfunctions. Several signaling cascades have been implicated, affecting processes as varied as protein processing, protein expression, and subcellular protein localization, among others. Therefore proteomic profiling of phosphorylated proteins in clinical specimen is thought to be getting more important tool to detect abnormality in patient tissues. Two-dimensional gel electrophoresis-based proteomics is the most appropriate for analyzing protein phosphorylation because phosphorylated protein shows drift in isoelectric point in the first dimension isoelectric focusing. And phosphorylation is easily detected by various methods including autoradiography, immuno-blotting and in-gel staining with Pro-Q Diamond fluorescent dye. Phosphorylation can be confirmed by detecting neutral loss by MALDI-TOF MS in post-source decay (PSD) mode. We tried to profile phosphorylated protein human brain and human neuroblastoma cell line SH-SY5Y. Phosphoprotein specific staining with Pro-Q diamond was followed by SYPRO-Ruby staining and superimposed those two patterns in a Dual Color Display window of PDQuest . Both phosphorylated and unphosphorylated proteins were identified by paptide mass fingerprinting and phiosphorylation was confirmed. Thus obtained data of phosphorylated proteins in human brain tissue and human neuroblastoma SH-SY5Y cells.
抄録全体を表示
-
荒木 令江, 長 経子, 平野 穣, 小野 聡, 古石 和親, 川野 克己, 戸田 年総, 荒木 朋洋, 次田 晧, 福永 浩司, 佐谷 秀 ...
セッションID: 1G4-3
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
The brain hippocampus region has been known to possess markedly higher susceptibility to ischemic hypoxia and proceed to the specific neuronal apoptosis. Recently, relevance of p53 tumor suppressor gene to the induction of neuronal cell death, especially in central nervous system (CNS), has been focused. To elucidate the mechanism of the specific p53 dependent/independent apoptosis in hippocampus neurons, we established the mouse model induced neuronal apoptosis in hippocampus regions by bilateral common carotid artery occlusion (BCCAO), and analyzed the specific hippocampus proteins related to the ischemic apoptosis, using p53 gene knockout mice (p53-/-) comparing with those of p53 wild mice (p53+/+), by two proteomic strategies; 1) Differential display (comparative 2DE) by 2D-DIGE (fluorescence difference 2-D gel electrophoresis technology), 2) Differential display (comparative LC-MASS) by cICAT (cleavable isotope-coded affinity tags) method. In 2D-DIGE, about 4450 protein spots were detected in total, and 213 spots were found as ischemic stress sensitive or p53 specific proteins, such as p53 specific but ischemic apoptosis independnt;93, p53 independent but apoptosis dependent;53, p53 and apoptosis dependent but these factors are not interacted;14, and p53 and apoptosis dependent and these factors are interacted;39 (p53+/+specific apoptosis related;24, p53-/-specific apoptosis related;15). On the other hand, in cICAT method, 722 proteins were identified in 1074 unique peptides in 16000 peptides analyzed from hippocampus, and 467 (216 increased and 234 decreased) proteins were identified as the p53 specific apoptosis related proteins. These unique proteins identified by both methods included proteins related to apoptosis and cell cycle, redox related proteins, structure proteins, primarily chaperone proteins, kinases, small GTPases, cellular metabolism enzymes, and unknown proteins. These data suggest that the neuronal ischemic apoptosis in mouse hippocampus results from complex interactions between p53 dependent pathways involving redox enzyme reduction, induction of stress-induced proteins and apoptosis/cell cycle related proteins, and p53 independent pathway involving increased metabolism, disruption and reproduction of membrane and structural proteins, and ,ultimately, cell integrity breakdown. The prospective ability of those strategies for functional study of the neuronal apoptosis in central nervous system will be discussed.
抄録全体を表示
-
佐藤 雄治, 島崎 早矢香, 石田 麻沙美, 戸田 年総, 山本 晴彦, 遠藤 玉夫
セッションID: 1G4-4
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Neurofibrillary tangle is one of the hallmarks of Alzheimer's disease (AD) and is composed of paired helical filament (PHF) of cytosolic protein tau. Previously we reported the occurrence of N-glycosylation of PHF tau prepared from the AD brain and we found that structures of N-glycans of PHF tau were mainly high-mannose type (Sato et al. 2001 FEBS Lett. 496, 152-160). N-glycosylation of PHF tau was thought to be responsible for the maintenance of PHF structure (Wang et al. 1996 Nat. Med. 2, 871-875). These results suggest that AD may be related to aberrant N-glycosylation of tau and that cytosolic protein may be abnormally N-glycosylated in AD brain. Because the aging is one of the risk factors of AD, we examined whether N-glycosylated proteins of brain cytosol increased in the aged process. We analyzed glycoprotein-expression level in the brain soluble fraction by 2D-PAGE in combination with concanavalin A (Con A) staining. In cortex, it was found that eight spots in aged rat (30-month-old) were more reactive to Con A than in young-adult (9-week-old). Among them, reactivations of three spots in the aged-rat were also observed in hippocampus, cerebellum and spinal cord. These results suggested that the increase of Con A-reactivity of soluble proteins was the common phenomenon in the central nervous system. Because the reactivations were diminished by Endo H digestion, these glycoproteins had high-mannose type N-glycans. However, the appearance of cytosolic N-glycosylated proteins in the aged brain is unclear because N-glycosylation and its processing proceeded in the lumen of endoplasmic reticulum (ER) and Golgi apparatus. Recently it was reported that misfolded N-glycosylated proteins were degraded by ER associated degradation (ERAD) system. If the deterioration of ERAD occurred in the aged rat, N-glycosylated proteins may be accumulated in the cytosol. We are now trying to identify these accumulated glycoproteins.
抄録全体を表示
-
奥泉 盛司, 剣持 聡久, 佐藤 雅男, 高木 陽子, 上條 憲一, 次田 晧
セッションID: 1S1-1
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
In the post-genome-sequence era, both the amount and the variety of proteome data are rapidly increasing due to the progress of protein identification methods such as mass-spectrometry (MS), high-throughput liquid chromatography. To manage a wide variety of enormous data and control the quality of the analysis results, IT platform for proteomics data is necessary for the researchers. We have developed a proteomics solution system which consists of a sample management system by using barcode tracing, a MS spectrum data archiving system, a proteome analysis result management system, a data mining system of related papers from literature databases and so on. This IT platform can facilitate proteomics researchers exploring relationships between sample resource information, MS data and identification results. We have proposed an XML format, HUP-ML (Human Proteome Markup Language), for proteomics databases, and for exchanging proteome data. HUP-ML can describe the preparation parameters as well as the results of 2D gel electrophoresis analysis / LC analysis by adopting a proteome-analysis-oriented data model, which would make proteome data comparable and reproducible. HUP-ML involves such information as sample preparation, 2D gel electrophoresis conditions, spot identification results, amino acid sequences. We provide the editor with graphical user interface (GUI) to create and utilize XML formatted proteomics data. In addition, we developed the database of human kidney glomerulus proteome using by 2-DE analysis. The database is accessible through the Internet and it exemplifies the way of proteome data exchange by using the XML format and its contribution to collaboration between researchers.
抄録全体を表示
-
森澤 拓, 久富 寿, 廣田 三佳子, 戸田 年総
セッションID: 1S1-2
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
The TMIG (Tokyo Metropolitan Institute of Gerontology)-2DPAGE has been improved to achieve efficient proteome data exchange among collaborative research community. We developed an integrated XML viewer and the XML database system (PHP + PostgreSQL) for 2DPAGE. And we further optimized our TMIG-XML format for data sharing in the user community. The format is designed with 2 layers of the two-dimensional electrophoresis gel information and spot information in the gel images. It has the locale information of the spots. The clickable imagemap functions for the representation of the spot information are both available in the XML database system with PHP and in the viewer with Object Pascal (Delphi 7.0). Individual spots on an experimental gel image are easily matched to corresponding spots on our standard gel image of our TMIG-2DPAGE proteome database using the viewer. The viewer must be the efficient collaborative tool in the user community. The viewer and the database system both allow the user to click on the spots on the gel image to obtain the appropriate entry in the major sequence databases. Moreover the TMIG-2DPAGE database client and the private TMIG-2DPAGE in the TMIG's intranet as the simple laboratory information management system will be constructed. The additional format with the project information, the worker information and time-stamp of major items will be imported to the expanded TMIG-XML format.
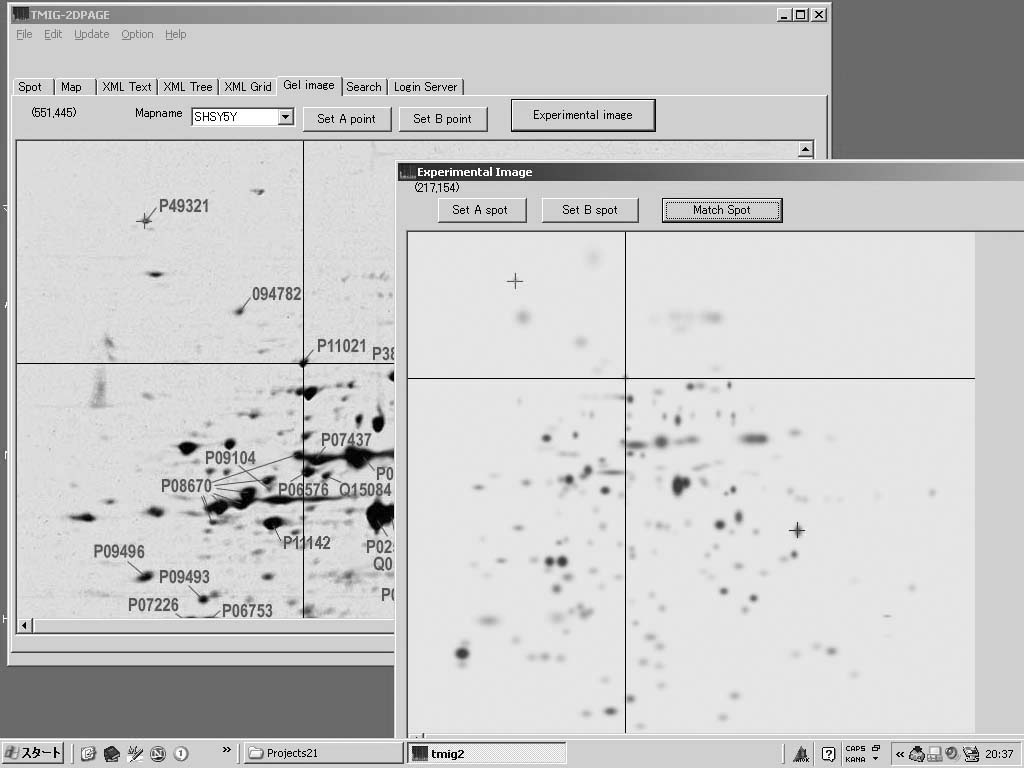
抄録全体を表示
-
吉田 豊, 許 波, 張 蛍, 宮崎 賢司, 佐藤 雅男, 奥泉 盛司, 剣持 聡久, 上條 憲一, 次田 晧, 大澤 哲雄, 矢尾板 永信 ...
セッションID: 1S1-3
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
We have analyzed proteins expressed in the glomerulus of normal human kidney by 2-DE and identification through MALDI-TOF MS and/or LC-MS/MS. Glomeruli were highly purified from kidney cortices with normal appearance obtained from patients under surgical nephrectomy due to renal tumors. The glomeruli were separated by 2-DE conducted with 25x20 cm 12.5 % separation gels coupled with IPG strips of pH range of 3-10, and silver-stained. From 2-DE gels of 5 subjects with no apparent pathologic manifestation, a synthetic gel image was produced by using PDQuest. Nearly 350 protein spots were so far identified, which were grouped into 18 lager categories on the basis of Gene Ontology terms. Although most of proteins identified include cell structural proteins, metabolic enzymes, and protein metabolism, significant number of proteins implicated in signal transduction (25), cell cycle and proliferation (10), and stress response (13) were also identified. Thus, our database might be useful for elucidating biological processes altered under different physiological or pathological conditions, although identified proteins were only a fraction of all the proteins expressed in the glomerulus. In order to improve proteomic approach for the glomerulus, we have undertaken analysis to specify proteins abundantly expressed in the glomerulus of normal kidney, and have already detected 204 proteins preferentially expressed in the glomeruli. A database of normal glomerular proteome are now being constructed by an XML-based editor (HUP-ML) designed for construction of proteome database. The database includes annotations such as protein name and synonyms, accession number of protein database, observed and theoretical pI and Mw, accession number of cDNA database, gene name, GO classifications, and other probable candidates or co-migrated proteins. The database will be submitted on a Web site for public access.
抄録全体を表示
-
佐藤 陽美, 福田 美紀, 重高 誠, 若松 容子, 井上 陽子, 溝口 佳伸, 富岡 伸夫, 板井 昭子
セッションID: 1S1-4
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
We developed a comprehensive information platform for bioscience research and drug discovery in the post-genome era, named
KeyMolnet.
Thanks to the technological advances of recent years, it is now possible to obtain very large quantities of data at a time. The DNA microarray technology might enable us to do so in the easiest way in the present time. However, it is very difficult to clarify biological function only by gene expression data even if they were cyclopedic. It needs information on proteins and also needs to integrate various kind of biological information. A platform for extracting meaningful information from such large quantities of data and for analyzing the relationship between the data and biological function, has long been awaited. We therefore developed an advanced tool, which can generate molecular networks upon demand, and can bind them to physiological events, medical and drug information.
Using
KeyMolnet, we show new approaches to network analysis for proteomics and genomics research.
KeyMolnet enables practical approaches to research into biological mechanisms, which in turn contributes to not only proteomics research but to discovery in medical, pharmaceutical and life sciences.
抄録全体を表示
-
田之倉 優
セッションID: 1S2-1
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
In 2002, RIKEN and eight university consortiums started on the National Project on Protein Structural and Functional Analyses of Japan (Protein 3000). Our consortium focuses on structural genomics on development and differentiation of organisms and replication and repair of DNA. We plan to determine more than 70 protein structures in 5 years starting at April 2002, and aim to elucidate general molecular mechanisms of cell division, cell differentiation, cell communication and cell death as well as DNA replication and DNA repair. Our consortium comprises 23 members and 6 cooperators who belong to 14 universities, 2 national institutes, and 3 companies in Japan and are divided into 4 groups according to their research fields, i.e., overproduction of recombinant proteins and their functional analysis, technical support for overproduction, three-dimensional structural analysis, and bioinformatics. In addition to the conventional techniques, we take advantage of new recombinant protein production systems such as the cell-free protein expression system using wheat germ extract developed by Prof. Yaeta Endo (Ehime Univ.) and the cspA (cold shock protein A) promoter-based E. coli expression system (Takara Bio Inc.).
In the first two years to March 2004, we chose more than 500 target proteins from thermophilic archaea, fruit fly, mouse, human and so on. We determined 84 protein structures, of which 44 were deposited into PDB, filed 15 applications for patents, and published 188 peer-reviewed papers. In this workshop, I am going to talk about our strategy in structural genomics and some results obtained thus far.
抄録全体を表示
-
田仲 昭子, 横山 茂之
セッションID: 1S2-2
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
The goal of structural proteomics is to reveal the genome-wide relationships between protein structure and function. To pursue this grand goal, we have been entrusted with the
National Project on Protein Structural and Functional Analyses (Protein 3000 Project) in collaboration with many Japanese scientists.
We theoretically and computationally select target proteins with important biological functions and disease-related functions, and then examine their expression and solubility by our unique cell-free method. Hundreds of the selected proteins have been prepared on a large-scale. Using NMR spectroscopy or X-ray crystallography, our group determined more than 200 structures of proteins, mainly derived from mouse and human, in the 2003 fiscal year. In addition to determining the three-dimensional structures of proteins, we also analyze various molecular functions, including protein-DNA or protein-protein interactions and protein-ligand affinity. We are developing and applying the technologies of
in silico ligand screening to study the functions of target proteins. These functional results provide important clues for selecting the next target proteins to study.
To transfer our results to industrial R & D, we have begun a new drug discovery collaborative program, the
Partnership Program. Under the basic collaborative research agreement, we instantly disclose the protein-research progress-reports to partner companies, which then select target proteins for subsequent individual collaborations. Under the individual collaboration research agreement, we provide protein samples and information for further research to the partner companies. This partnership will provide the basis for the next-generation of theoretical drug discovery in Japan.
抄録全体を表示
-
藤吉 好則
セッションID: 1S2-3
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
A biological cell, such as a nerve cell regulates a cell signalling mainly by ion channels. Water movement through membrane should therefore be strictly separated from the movement of ions. This means water channels must be highly specific for water to prevent any ions. The studies on function of a water channel, aquaporin-1 for the last decade put us some puzzling questions. For answering the puzzling questions, structure of aquaporin-1 was analysed at a resolution of 3.8 Å by one of the third generation cryo-EM and electron crystallography. We also analysed a structure of another water channel expressed in brain. The muscle-derived electric organ of the
Torpedo electric ray is highly enriched in acetylcholine (ACh) receptor-containing membranes which have been a major source of material for structural study of the receptor. The membranes are readily converted into tubular crystals, having helical symmetry. By imaging of the tubular crystals in thin films of amorphous ice, structure of ACh receptor was analysed at a resolution of 4 Å.The ACh receptor has a cation-selective pore, delineated by a ring of five subunits. In each subunit, four membrane-spanning segments, M1-M4 were predicted and the second membrane-spanning segment, M2, shapes the lumen of the pore and forms the gate of the channel. The gate is a constricting hydrophobic girdle at the middle of the lipid bilayer, formed by weak interactions between neighbouring inner helices. When ACh enters the ligand-binding domain, it triggers rotations of the inner parts of the domains of α-subunits. These rotations are communicated through the inner (M2) helices and open the pore by breaking the girdle apart. We analysed structure of voltage sensitive Na
+-channel as well as IP
3 receptor by single particle method. By utilizing our cryo-EM we have now good chance to understand functions of channels from structural point of view.
抄録全体を表示
-
武内 桂吾, 野崎 学, 根本 直, 山根 國男
セッションID: 1S2-4
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Bacillus subtilis is a Gram-positive bacterium having high levels of ability to secrete extracellular enzymes and its genome sequence was published at 1997 by Kunst
et al. (1).
B. subtilis possesses SecA protein, which is a prokaryote unique ATPase to plays a central role on the extracellular enzyme translocation across the cytoplasmic membrane. However, the relation between SecA function and the localization of membrane proteins in
B. subtilis is studied very little. To resolve this subject, the ideal method, in which the
de novo synthesized proteins were labeled with
15N and the relative amount of newly located proteins into the cell membrane were quantitively analyzed by MALDI-TOF MS after the separation of the proteins by 2D-PAGE, provided an efficient one. As results, the localization of 25 proteins in the tested 28 was inhibited by the absence of SecA. The dependency of the 28 proteins on SecA for their localization into the cell membrane could be classified into three groups by the strength of the inhibition. The first protein group including all lipoproteins and some membrane proteins was directly dependent on SecA, whose localization was immediately inhibited by the lack of SecA. In contrast, the second protein group including most of integral membrane proteins was indirectly dependent on SecA because the inhibition by the lack to SecA was delayed. Third group was independent from SecA because no inhibition of the localization was observed. These results suggested that several kinds of machinery relating to the membrane protein localization will be equipped in
B. subtilis cells.
(1) Kunst, F. et al., Nature 390. 249-256 (1997).
抄録全体を表示
-
志村 清仁, 脇 卓真, 岡田 政喜, 戸田 年総, 笠井 献一
セッションID: 1S3-1
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Analysis of specific interactions between biological molecules is a key element in functional proteomics. Capillary electrophoresis has been used to analyze molecular interactions in free solution on the basis of the mobility change caused by the formation of complexes. The mobility of a molecule is sensitive to the changes in its electric charges, which set this method against others for the analysis of interactions. We have worked to establish robust protocols for successful applications for a wide range of interactions using an electrophoresis instrument (SpectruMedix HTS 9610) equipped with 96 fused-silica capillaries and a laser-induced fluorescence detector and have succeeded in developing two important functional tools for the instrument. The first is the mobility moment analysis that enables direct comparison of the electropherograms obtained in different capillaries and determination of dissociation constants. The second is the dual buffer system that uses an electrophoresis buffer of pH 9.35 and a sample buffer of pH 7.35 permitting the analysis of interactions at physiological pH within a 15 min cycle time with stable base lines even with bare-silica capillaries. Flanking of a sample plug with sample buffer plugs was effective to detect relatively weak interactions. Interactions between antigen-antibody, lectin-glycoprotein, enzyme-inhibitor and lectin-sugar were analyzed. In the case of lectin-sugar interactions, the mobility change caused by interaction was amplified using an affinophore, a sugar ligand attached to an anionic polymer, succinylated polylysine (SPL). Competition analysis revealed a spectrum of sugar specificity of a lectin with good agreement with the previous results obtained with different methods. Multiplexed capillary electrophoresis instruments seem to be well worth adding in an analytical repertoire for biomolecular interactions, providing dissociation constants that are essential to characterize interactions.
抄録全体を表示
-
平野 久, Tan Jian-zhong, Suzuki Nobutake, Arima Mikiko, Oba Mitsuyoshi, K ...
セッションID: 1S3-2
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Since proteins interact with other proteins to perform their particular cellular task, analysis of the protein-protein interaction is important to determine the function of proteins. Protein-protein interaction has been analyzed by several methods such as yeast two-hybrid system, immunoaffinity purification technique and protein chip. Among them, protein chip has been considered the most promising tools for high-throughput analysis of protein-protein interaction at protein level. In the conventional technique, proteins expressed by DNA (RNA) or natural proteins have been purified and immobilized on the chemically modified glass plate or membrane to produce the protein chip. However, purification of the proteins has been laborious and time-consuming, and the purification of a number of proteins has been often impossible. This is a limiting factor to perform the high-throughput analysis of protein-protein interaction. We developed a technique for high-throughput analysis of the interaction using a novel protein chip. In this technique, proteins are separated by gel electrophoresis, and electroblotted onto the diamond-like carbon coated (DLC) stainless steel plate, of which surface is modified with N-hydroxysuccinimide ester, to produce a high-density of protein chip. Proteins in the gels can be immobilized covalently on the DLC plate with high blotting efficiency (30-70 %). Proteins extracted from the cells were probed with the proteins on the DLC plate, and the interacted proteins were detected by matrix assisted laser desorption ionization (MALDI)-time-of-flight mass spectrometry using the plate as a MALDI sample target. This technique has a great potential of high-throughput analysis of proteins interacted with thousands of proteins separated by two-dimensional gel electrophoresis.
抄録全体を表示
-
根本 直, Fedorov Dimtri, 古明地 勇人, 金澤 健治, 上林 正己, 北浦 和夫
セッションID: 1S3-3
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
The large-scale structural proteomics projects have been launched recently and are aggressively undergoing, therefore the amount of protein structural data has been increasing at a considerably high-speed.
Normally, thee dimensional structures in the database(s) are produced by Molecular Dynamics calculation (MD) or Simulated Annealing(SA) methods by computer at their final stage starting from raw X-ray or NMR data sets. While we look at these attractive structures, they are still rough to analyze and estimate energy.
A new methodology for macromolecules named Fragment Molecular Orbital (FMO) method has been proposed for the calculations of structures and properties based on non-empirical electronic structure calculations [Fedorov et. al.,
J. Chem. Phys. in press]. FMO is applicable to enormously large numbers of atoms in biological systems than conventional Molecular Orbital method. Furthermore, Komeiji and co-workers have applied FMO to MD simulations: FMO-MD which makes possible to perform MD simulations without force fields [Komeiji et. al.,
Chem. Phys. Lett., 372, 342-347, 2003].
An example for ligand-receptor interactions, we analyzed a
Bombyx mori sex pheromone binding protein (BmPBP) with its ligand (bombykol) which structures have been determined both by X-ray crystallography [Sandler et al.,
Chem. Biol. 7, 143-151, 2000] and by NMR [Lee et al.,
FEBS Letters 531, 314-318, 2002]. Starting from the experimental structure, we optimized the structure of the ligand and entire binding pocket in the ligand-protein complex. The electronic charges, interaction energy and binding energy was calculated and estimated in atomic order. The binding mechanism will be discussed based on the interaction energies. Several other examples of FMO calculation will be presented.
抄録全体を表示
-
S.J. Lynden, O.C. Idowu, P. Periorellis, M.P. Young, P. Andras
セッションID: 1S3-4
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
The aim of our work is to accumulate protein interaction data and to develop computational techniques for analysing the topologies of protein interaction networks to reveal network vulnerabilities. We have developed a variety of network analysis algorithms which aim to discover network vulnerabilities, consisting of small sets of proteins, which may be used to aid the discovery of pharmaceutical drug targets for fighting antimicrobial infections. The developed algorithms include the recognition of highly connected [hub] nodes, [bottleneck] nodes which connect network sub-clusters, and the analysis of nodes which participate in topological structures such as clusters, pathways and loops. A composite database has been constructed which combines the complete sets of protein interaction data available from the Database of Interacting Proteins (DIP), the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) and Kyoto Encyclopaedia of genes and Genomes (KEGG) Pathway database. Following an analysis of the Bacillus subtilis protein interaction network, it was found that 40% of the protein targets predicted by our techniques are encoded by genes known to be essential for the survival of the organism, therefore supporting the effectiveness of our approach. Further work will involve the analysis of other bacteria and the development of new graph theoretic techniques for network analysis.
抄録全体を表示
-
夏目 徹
セッションID: 1S3-5
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
One of the strategies of functional proteomics, research aiming to discover gene function at the protein level, is the comprehensive analysis of protein-protein interactions related to the functional linkage among proteins and analysis of functional cellular machinery to better understand the basis of cell function. One of the most powerful means for the identification of proteins in cellular compartments or macromolecular complexes is liquid chromatography (LC) based electrospray tandem mass spectrometry (MS/MS). To enhance the sensitivity and resolution of MS/MS analysis, we have developed a novel direct nano flow LC-MSMS system (DNLC-MS/MS). This system enables us to identify 100-150 proteins in a single run at low femtomole level without pre-separation of each component by 2D or 1D-electrophoresis. By this approach, protein components in even large macromolecular complexes can be comprehensively identified in a high-throughput and automated manner.Systematic efforts to understand protein-protein interactions are under way in our research project using human full length cDNAs. In the project, the cDNAs were fused to an epitope tag sequence and expressed in human cells. The expressed tagged proteins (bait) together with their associating partners (prey) were purified from cell lysate using anti-tag affinity beads. The isolated protein complexes were identified using the DNLC-MS/MS system. So far, approximately 2000 analyses (800 human cDNAs) have been performed. Supported by NEDO (New Energy and Industrial Technology Development Organization).
抄録全体を表示
-
古田 大
セッションID: 1S4-1
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
The main stream in the proteomics field today is the identification of proteins resolved in two-dimensional electrophoresis, by performing in-gel protein digestion and the peptide mass fingerprinting (PMF). In that case, the entire sample on a protein spot is sacrificed for just one analysis. The Chemical Inkjet Printer (ChIP) is a revolutionary new approach to PMF and protein macroarray analysis. The ChIP reproducibly dispenses picoliter volumes of reagents to defined locations, only tiny areas in the protein spot, opening new horizons for microscale protein research. Protein samples are to be processed by the ChIP after blotted onto PVDF membrane from the gel, that is, proteins on the membrane are successfully digested and MALDI-TOF mass spectra can be obtained directly from the membrane (see Figure). Then microscale on-membrane protein identification and characterization can be rapidly generated bypassing time- and sample-consuming procedures of in-gel digestion, peptide extraction and desalting steps. The technology can also rapidly microdispense multiple antibodies to screen for antigens and achieve minute-scale Western blotting. In this session, key features of the ChIP and its applications in biochemical sciences will be discussed.
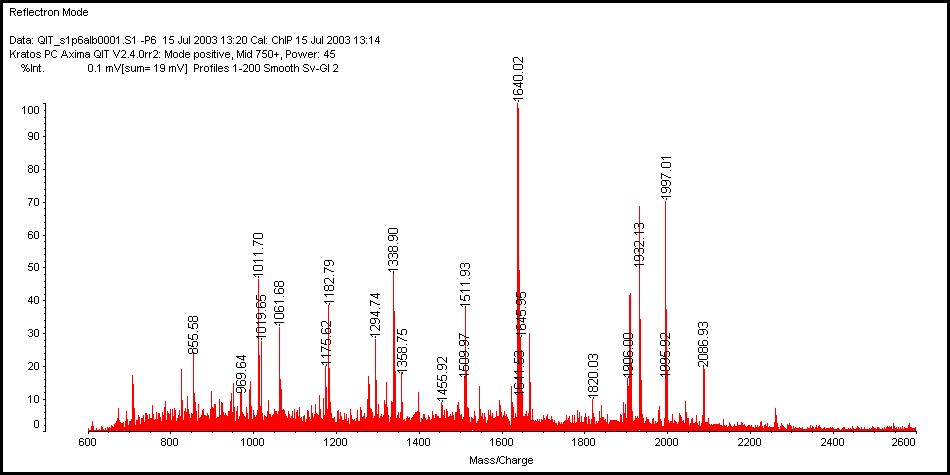
抄録全体を表示
-
大房 健, 山縣 彰, 吉里 勝利
セッションID: 1S4-2
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Protein phosphorylation is one of the most important post-translational modification of proteins and plays a variety of biological processes such as differentiation, cell division, metabolism, cell-cell communication, and disease. The phosphorylation of a protein cannot be predicted from its DNA sequence, and is determined only by analyzing protein itself. Proteome analysis using 2D gel electrophoresis is a most powerful technique for solving the composition of total proteins contained in cellular proteins. Although several separation techniques of proteins has been employed in proteome analyses, one of the most common technique is two-dimensional electrophoresis which simultaneously resolve several thousands of proteins including protein variants produced by the co- or post-translational processing such as phosphorylation, glycosylation, and degradation. We devised a new method (phosphoproteomics) to comprehensively map phosphoproteins on 2-D gels by comparing gel images of phosphatase-treated and non-treated samples.
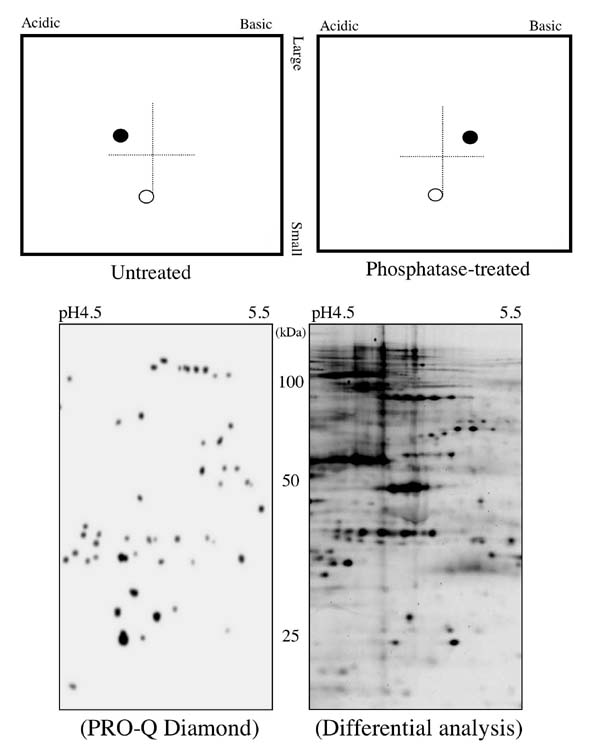
抄録全体を表示
-
宮崎 賢司, 上條 憲一, 次田 晧
セッションID: 1S4-3
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Protein in polyacrylamide gel was successfully C-terminally truncated with use of acetic anhydride formamide solution and its C-terminal amino acid sequence was analyzed by measuring masses of a series of truncated products with MALDI-TOF MS in both positive ion mode and negative ion mode. As a model protein in gel, CBB stained 5 ug myoglobin was subjected to the truncation reaction. After dehydration of the gel by acetonitrile, the gel was put in 30% acetic anhydride formamide solution and reacted at 50˚C for 110h to truncate the C-terminal amino acids succeesively. Then, hydration reaction with 10% dimethylaminoethanol aqueous solution at 60˚C for 1h was employed to obtain clearer mass spectrum from the truncated protein. The hydrated product was digested with trypsin in gel and the extracts were subjected to MALDI-TOF MS. Negative ion mode was found to be more preferable to obtain the C-terminal sequence information in this method. In the analysis, up to 5 residues truncated products ions were observed in the mass spectrum.
抄録全体を表示
-
中西 洋志, 中川 将利
セッションID: 1S4-4
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
There are many peptides and proteins whose amino acid sequence cannot be analyzed, because full fragmentation ions are not detected by PSD method in MALDI- TOF- MS.In our proteome researches, antigen peptides produced by digestion of nucleic protein of influenza virus were in the case. Sequences of the amino acid residues in peptides modified in thiol group of N-terminal cysteine by fluorescene were completely analyzed with sufficient fragment ions by PSD method. Similar analyses of antigen peptides with cysteine residue at C-terminal or non-terminal positions were performed. Sufficient numbers of fragment ions for the peptide sequencings were detected in MS spectra and the sequence analyses were perfectly made.Furthermore, modifications were also made for N-terminal position of other general peptides without cysteine residue. Similar modification reaction gave satisfactory results of MS analyses of the peptide sequencings.Since there is the possibility that amino group at the side chain of amino acid residue is also modified by the fluorescene along with the N-terminal position, we examined reactivity of the fluorescent reagent against an amino group at side chain of arginine, asparagine, glutamine, or lysine residue in peptides to conform the useful common application of the method to all peptides. The obtained MS spectra clearly showed that all modified peptides gave completely full fragment ions for the sequence analyses. The modification by fluorescent reagent to general peptides is a complete and reliable technique for the sequence analysis of peptides by using MALDI-TOF-MS PSD method in proteome research.
抄録全体を表示
-
石濱 泰, 小田 吉哉, 田畑 剛, 宮本 憲優, 相根 康司, 佐藤 俊孝, 長洲 毅志
セッションID: 1S4-5
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Metabolic labeling for quantitative proteome has been developed using cultured cells with different states. Because the incorporation of stable isotopes into proteins were accomplished at the initial stage of the sample preparation using isotopically-modified media, metabolic labeling approaches have a significant advantage over the chemical labeling approaches where some variations between samples can be introduced during preparation. However, samples from tissues are difficult to label metabolically. In this presentation, we will show a new approach for quantitative tissue proteomics using cultured cells as sources of internal standards for tissue samples.
After homogenizing the brains from wild type and ADAM22 knockout mice, BISCUIT cells were added into each sample. Firstly, reproducibility was demonstrated using cytosolic malate dehydrogenase (cMDH), which can be enriched with affinity purification. Five independent trials resulted in low variance (CV=0.041), and linearity of estimation curve resulted quite high (R2=0.97), indicating that the novel quantification using BISCUIT cells could be applicable for comprehensive analysis. To prove the significance of the method, proteins from hippocampus sample treated with kainate were compared with non-treated counterpart. It should be also emphasized that absolute quantification could be achieved in case the protein amount was determined in BISCUIT cells in advance. Actually we examined the protein amount of cMDH, HSP60, HSP70, HSP90, 14-3-3, and myosin by using antibody, enzyme assay or synthetic peptides, resulting the estimation of absolute amount of these proteins in mouse brain. In summary, we have developed a general method for simultaneous identification and quantification of tissue proteome with high precision. The procedure should be applicable to variety of cell systems and tissues, so that BISCUIT cells might be the global standard for in vivo labeling, quantitative proteomics study.
抄録全体を表示
-
細川 桂一, 高 嘉凌, 鍋谷 卓司, 次田 晧, 宮崎 賢司, 上條 憲一
セッションID: 1S4-6
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Genomic coding plays a fundamental role in determining hereditary traits of all living organisms. One the other hand, in higher organisms, epigenetic changes in chromatin structure, which include DNA methylation and histone modifications give rise to inheritable changes in phenotypic expression of the genes during cellular differentiation, development as well as pathological changes in cells such as cancers.Above all, chemical modifications (acetylation, methylation, phosphorylation and ubiquitination) of nucleosome core histones H2A, H2B, H3 and H4 play a crucial role in epigenetic requlation of genes. Here we report a simple method to characterize and localize the modifications in the four core histones.Liver tissue was homogenized and extracted with 0.165M HCl-0.165M NH
4Cl. The centrifuged clear supernatant containing histone mixture was subjected to 1D SDS-PAGE. Each separated histone bands except histone H2B was excised out of gel and subjected to succinylation in acetonitrile/formamide, followed by trypsin digestion. The peptides were analyzed by MALDI-TOF MS. Histone H2B was directly subjected to in-gel-digestion with lysylendopeptidase without succinylation.These data will determine the loci and modification of amino acid residues in the histones, which reflect the mode of epigenetic control of cells and serve as a strong tool for clinical diagnosis.
抄録全体を表示
-
藤井 清永, 西村 俊秀
セッションID: 2G1-1
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Blood plasma is not only the primary clinical specimen but also represents that the largest and deepest version of human proteome present in any sample. In clinical and diagnostic proteomics for human plasma, it is essential to develop a comprehensive and robust system for a large-scale proteome analysis. Recently, multi-dimensional liquid chromatography/ tandem mass spectrometry (LC/MS/MS) systems have been developed as powerful tools especially for protein identification of highly complexes and have achieved resolution power compatible to two-dimensional gel electrophoresis. However, these systems need to be further developed in terms of ease in usage and industrial applicability to the routine clinical use, because their application to clinical researches requires an analysis of a large number of human samples with stability and reproducibility. Therefore, we have constructed a technically simplified and high-speed/throughput LC/MS/MS system with micro-flowing reversed-phased chromatography and linear ion-trap mass spectrometry equipped with nanoelectrospray interface. Additionally, the system was combined with on-line strong cation exchange chromatography as a multi-dimensional protein profiling system. For a large-scale human plasma proteome analysis, we have also established protein depletion, in-solution digestion and data-integrating/mining systems with an automated operation. Here, we will present the application of our fully automated multi-dimensional protein profiling system to plasma proteome analysis for clinical proteomics.
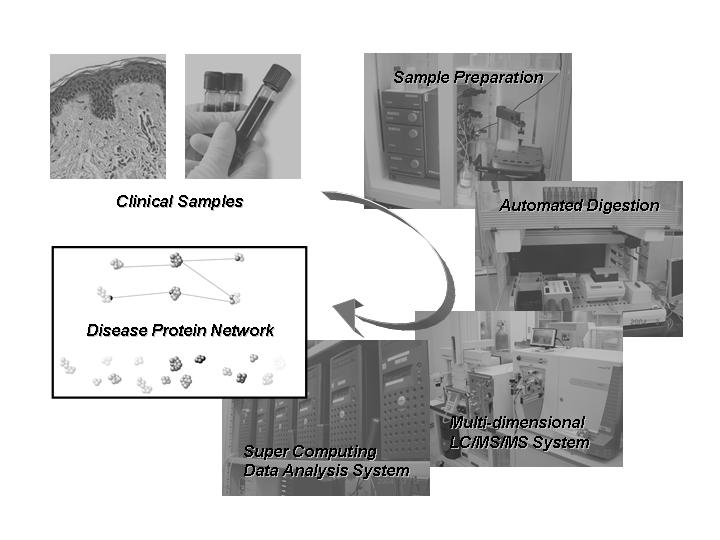
抄録全体を表示
-
Michael H. Simonian, Edna Betgovargez
セッションID: 2G1-2
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
An ultimate objective of proteomics is the understanding of complex biological systems, which can lead to new diagnostics and therapy. The first step toward this end is the profiling of protein differences between states of a proteome. One approach to proteome profiling fractionates the proteome into intact proteins with subsequent analysis for structural characterization and identification of the protein differences. This paper presents a multidimensional approach for proteome profiling that utilizes two-dimensional liquid chromatography for fractionation of the proteome followed by analysis with capillary electrophoresis (CE) and/or mass spectrometry (MS). The first-dimension separation is done by chromatofocusing over a pH range from 8.5 to 4.0, which separates proteins by pI. Fractions are collected based on pH intervals as detected by a pH monitor. The proteins are detected by UV absorbance at 280 nm. The first-dimension fractions are separated in a second dimension by high-resolution, reversed-phase chromatography with a trifluoroacetic acid - acetonitrile gradient. The proteins are detected by absorbance at 214 nm. Fractions collected from the second dimension are analyzed by CE and MS. This multidimensional fractionation and analysis approach was used to compare fasting and non-fasting human plasma. Glycoprotein content was analyzed by CE and proteins identified by MALDI-TOF MS. The removal of the six most abundant plasma proteins (albumin, IgG, transferrin, fibrinogen, IgA and IgM) with specific chicken IgY antibodies was evaluated for effects on the proteome profile. Distinct qualitative and quantitative differences in protein expression were observed between the fasting and non-fasting states of human plasma.
抄録全体を表示
-
Kathy Kozak, Feng Su, Kym Faull, Srinivasa Reddy, Robin Farias-Eisner
セッションID: 2G1-3
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
We have previously reported the identification of three ovarian cancer biomarker panels comprised of SELDI-TOF peaks representing fourteen differentially expressed serum proteins for the diagnosis of early ovarian cancer. We now report the identification and characterization of four of the proteins from the ovarian cancer biomarker panels. Using a variety of methods, including pI analysis, peak depletion studies, serum to pure protein comparisons, anion exchange fractionation, tryptic peptide analysis and LC-MS-MS, we confirmed the identity of two proteins that represent the m/z peaks 13.8 and 78.9 kDa in our original ovarian cancer biomarker panels. Furthermore, using western analyses and ELISA techniques (independent of SELDI), we confirmed the differential expression of transthyretin (13.8 kDa, prealbumin) and transferrin (78.9kDa) in a group of 44 test ovarian cancer serum samples. With the identification of these markers, we are now developing simple assays for improved clinical diagnosis and prognosis of ovarian cancer.
抄録全体を表示
-
藤ノ木 政勝, 亀森 哲, 深澤 一雄, 古野 元子, 山崎 龍王, 稲葉 不知之, 朱 坤, 香坂 信明, 太田 順子, 稲葉 憲之
セッションID: 2G1-4
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Surface-Enhanced Laser Desorption/Ionization (SELDI) ProteinChip® system is the new technology for the screening of biomarkers. This system is most fitting as an analysis system of sample obtained from patient since sample that it needs is very small amount.In the present experiment, we analyzed sera obtained from ovarian cancer patients in order to detect novel ovarian cancer markers and to establish the new diagnosis for ovarian caner. Sera of normal women and patients were analyzed by four types of chemical surfaces chip, which are hydrophobic (reverse phase chromatography), cationic (weak cation exchange), anionic (strong anion exchange) and IMAC (metal affinity chromatography) chip. In the preliminary protein differential display against two normal versus six patients, significant difference of protein expression were detected in analysis used three types of chemical chip, such as hydrophobic, cationic and IMAC chip. Therefore, we analyzed sera of sixty normal women and fifty two patients using these three chips. From the results of protein differential display in each chips, we detected many protein peaks that might be ovarian cancer marker. In the next step, four decision trees for diagnosis of ovarian cancer were constructed by the applied tree-based method by CART (Classification and Regression Trees) using the results of protein differential display in each chips. From results of the significant test for the decision tree constructed in the present experiment, it was suggested that each decision tree were the decision tree that did not pass over ovarian cancer patient.
抄録全体を表示
-
近藤 格, 岡野 哲也, 山田 昌代, 畠山 博充, 柿坂 達彦, 末原 義之, 山田 哲司, 広橋 説雄
セッションID: 2G2-1
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Cancer is the leading cause of disease death in Japan. Despite recent progress of early diagnostic modalities and therapeutic management, the long-term survival of cancer patients has been still poor and the extensive efforts to improve the prognosis have been devoted to cancer research. To identify the proteins associated with various clinical states of cancer and to utilize such proteins as diagnostic and prognostic biomarkers, we established the proteome analysis system based on two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) and bioinformatics approach. In our system, the quantitative protein expression profiles are generated by two-dimensional difference gel electrophoresis (2D-DIGE), in which protein samples are labeled with fluorescence dyes prior to 2D-PAGE. As high sensitive fluorescent dyes enable high-throughput laser microdissection, 2D-DIGE can achieve accurate clinical proteomic study. Multivariate algorithms and statistical-learning methods determine unique protein expression patterns corresponding to clinical features and mass spectrometric study identifies the proteins involved in the patterns. Proteomic study using cancer cell lines and clinical specimens of various types of cancer revealed that our proteome analysis system can identify the protein expression patters corresponding to 1) original organ and histology, 2) properties of metastasis and invasion, or 3) histological subtype of malignancies.
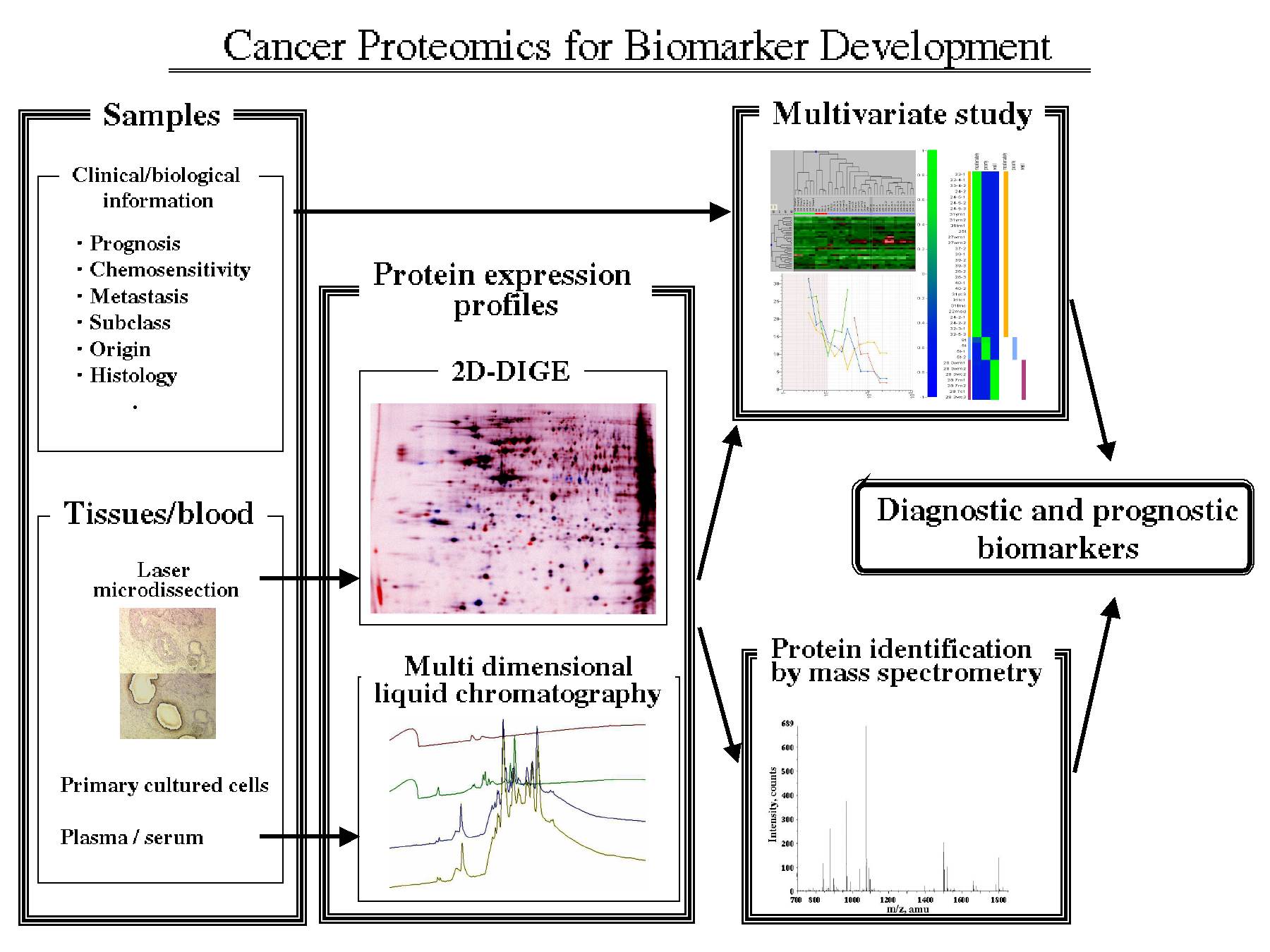
抄録全体を表示
-
荒木 令江, 長 経子, 戸田 年総, 古石 和親, 平野 穣, 小野 聡, 佐藤 陽美, 荒木 朋洋, 中村 英夫, 佐谷 秀行
セッションID: 2G2-2
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Oligodendrogliomas (OGs) are primary brain tumors, genetically characterized by chromosomal alterations in the 1p, 10q, 19q (LOH). Recent clinical trials have shown that anaplastic oligodendrogliomas (AOGs) possessing those LOH are sensitive to the chemotherapy and well controlled, yet AOGs without those LOH are insensitive and progressive, despite of their similar histological features. There is no information on the specific diagnostic markers or target molecules allowing for the recognition of OG cells in tumor tissues or understanding the sensitivity differences against the chemotherapy in two types of AOGs. To find out the molecular and genetic markers that aid in grading OGs and identifying patients with a better prognosis or response to chemotherapy, proteomic and transcriptomic differential analysis have been carried out between normal brain tissues and AOGs, or AOGs with and without the positive sensitivity against chemotherapy. Proteins and mRNA were extracted from the same brain tissues excised from AOG patients and subjected to the cleavable ICAT, 2D-DIGE, and DNA array analysis. By proteomics analysis, 331 proteins (82; more than 2 times increased, 231;less than 50 % decreased) were identified as tumor specific, and 58 proteins (28; increased, 30;decreased) were identified as specific for tumor with chemotherapy insensitive. In DNA array analysis, 998 transcripts were identified as tumor specific and 149 transcripts (111;increased, 37;decreased) were extracted as specific for tumor with chemotherapy insensitive being only 30% of similarity with the protein expression patterns, increased or decreased, observed in the proteomic analysis. These identified molecules contained not only those reported as tumor specific such as angiogenesis, adhesion, apoptosis, cell cycle related but also novel functional molecules in AOGs. The molecular signal networks of identified proteins were analyzed with KeyMolnet. The meaning of specific proteins in AOGs will be discussed.
抄録全体を表示
-
志和 美重子, 西村 由美子, 若田部 るみ, 大田 博俊, 加藤 洋, 矢守 隆夫
セッションID: 2G2-3
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
To discovery of new useful biomarkers for colon cancer, we performed Expression Difference Mapping analysis on 39 human cancer cell lines using SELDI ProteinChip platform. Several proteins were highly expressed in colon cancer cells (t-test, P<0.05), especially 12kDa protein showed the lowest p-value (0.0005) on SAX2. We then optimized the purification conditions for this candidate on chip, and directly transferred to conventional chromatography to purify. The purified protein was digested with trypsin or V8 protease to produce peptide mass fingerprinting analysis and collision-induced dissociation (CID)-tandem MS analysis with ProteinChip interface. Both identification results showed the candidate biomarker was a full-length of prothymosin-a. To evaluate whether prothymosin- a is a potential biomarker for colon cancer or not, we compared with the expression level of this protein between cancer cells and normal cells using real patients tissue samples. The peak intensity of identified prothymosin- a in colon cancer cells was clearly higher than that in normal colon cells in each patient. mRNA expression level of this protein in colon cancer was also higher than that in normal cells. Then we tried to capture prothymosin- a using antibody by Interaction Discovery Mapping analysis. The results of Interaction Discovery Mapping analysis was correlated with the results of Expression Difference Mapping analysis, it showed that the evaluation assay for this biomarker candidate could perform on SAX2 chip without establishment of antibody assay. These findings indicate that prothymosin- a could be a potential biomarker for colon cancer, and that the ProteinChip platform could rapidly perform the whole biomarker assay process from biomarker discovery stage to evaluation stage for the identified marker.
抄録全体を表示
-
岩立 康男, 堺田 司, 日和佐 隆樹, 滝口 正樹, 藤本 修一, 山浦 晶
セッションID: 2G2-4
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Gliomas are characterized by their highly heterogeneous aggressiveness even in the same histological category. The biological features of gliomas would be precisely described by global gene expression data at protein level. We examined the proteome of 93 surgical samples including 52 glioblastomas, 13 anaplastic astrocytomas, 10 astrocytomas, 8 oligodendrogliomas, and 10 normal brain tissue samples by using two-dimensional gel electrophoresis and mass spectrometry. On the hierarchical clustering developed from the proteome profiling patterns, the normal brain tissues and oligodendrogliomas were identified as distinct subsets. Furthermore, the proteome-based clustering significantly correlated with the patients'survival, and we could identify a biologically distinct subset of astrocytoma with aggressive nature. The discriminant analysis extracted a set of 37 proteins differentially expressed by the histological grading. Then, to identify the protein markers that are clinically useful for predicting efficacy of anticancer agents, we also investigated the correlation between the proteome profiling patterns and the in-vitro chemosensitivity measured by flow cytometric detection of apoptosis. The clustering map provided a particular distribution of chemosensitivity spectrum for each sample. We identified a set of 41 proteins that significantly affected the in-vitro chemosensitivity by discriminant analysis based on the proteome data. Many of the proteins that correlated with chemoresistance were categorized into the signal transduction proteins including the small G-proteins. The present study has shown that the proteome analysis is useful to develop a novel system for the prediction of biological aggressiveness and chemosensitivity. The proteins identified here could be novel biomarkers for survival prediction and rational targets for anti-glioma therapy.
抄録全体を表示
-
小寺 義男, 福冨 俊之, 古後 富久, 古館 専一, 大森 彬, 前田 忠計
セッションID: 2S1-1
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Technological advances in the proteomics has been quite remarkable. However, there remains difficulties in analyzing membrane proteins, peptides, minor proteins and functional state of proteins. In particular, peptides are hard to be detected with either two-dimensional electrophoresis or shotgun method. Furthermore, Such modification as oxidation and/or phosphorylation of peptides would surely affect their ionization efficiencies and quantitation of the modified peptides by mass spectroscopy would be quite difficult.The aim of our study is to search for peptides disease-specifically modified in the amount and functional states. Here, the peptides imply low molecular weight proteins including not only biologically active peptides but also cleaved ones. To achieve our purpose, we developed a method to successfully separate peptides in crude extracts of tissues with high yield and reproducibility. This method enables us to compare quantitatively amount of each peptide in normal and disease tissues. We applied the method to extraction of peptides in renal cortex of the diabetic (db/db) and its control mice (+/+). The peptides of three db/db mice were analyzed by MALDI TOF-MS, and their spectra were compared with those of three +/+ mice. The results showed that two peptides were changed in amounts due to diabetes mellitus. We have succeeded in purifying these peptides by two-dimensional HPLC and identifying them by amino acid sequencing.
抄録全体を表示
-
佐々木 一樹
セッションID: 2S1-2
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Peptidomics provides a powerful approach to identify peptide markers for cancer. Peptides that are preferentially secreted from tumor cells and stable in circulation could serve as ideal markers since determination of their serum levels may aid in cancer diagnosis and management. Thanks to developing proteomics technologies, studies on disease marker discovery are increasingly popular worldwide. However, endogenous peptides remain largely disregarded because they are not covered by current proteomics. It should also be noted that no genome-based approach is applicable to the discovery of peptide fragments associated with a particular tumor phenotype. In an attempt to identify potential peptide markers, we have initiated a mass analysis of peptides secreted by cancer cell lines and non-malignant cells grown in serum-free conditions. The fact that tumor markers currently used in clinics are secreted by cell lines points to the advantage of the use of cultured cells to this end. Our principal analytical platform rests on surface enhanced laser desorption ionization (SELDI) mass spectrometry for profiling and tandem mass spectrometry for identification. As proof of principle, samples from 75 cultured cells were analyzed on a SELDI mass spectrometer to identify candidate peptides associated with pancreatic adenocarcinoma only. A 3333 Da peptide, which occurred in 6 cell lines derived from pancreatic adenocarcinoma, turned out to be a COOH-terminal 29 amino acid fragment of the putative tumor suppressor protein DMBT-1. Studies using a specific antibody revealed that the peptide is secreted by growing pancreatic cancer cell lines but not by two non-malignant cell lines of pancreatic duct origin. Immunoprecipitation studies on surgical pancreatic adenocarcinomas also revealed that it is expressed in a cancerous lesion but not in a matched control lesion in 4 out of 5 cases.
抄録全体を表示
-
Per E Andren, Marcus Svensson, Karl Sköld, Anna Nilsson, Helena N ...
セッションID: 2S1-3
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Treatment of mice with MPTP is one of several models used to mimic Parkinson's disease (PD) in humans. First, we have developed a peptidomic approach to study a large number of neuropeptides and employed it to an investigation of the endogenous neuropeptide content of PD. Secondly, two-dimensional fluorescence difference gel electrophoresis (DIGE) was used to pinpoint significant differences in protein abundance. Finally, direct molecular imaging of the biological samples using MALDI-MS was shown to be a powerful tool for investigating the spatial distribution of peptides and proteins directly on tissue samples. The 2D DIGE analysis demonstrated significant changes in a number of striatal proteins following dopamine depletion using an animal model of PD. The peptidomics approach was developed using only mg quantities of brain tissue. The comparisons of neuropeptides revealed several differences in relative abundance both between the different animal groups and the different brain regions. The compared peptides comprise precursor families of both known secretory pathway proteins, including granin-like neuroendocrine peptide, CART, POMC, prosomatostatin, proenkephalin, and precursors with neuropeptides identified as novel. Further, our results show that MALDI imaging directly on tissue slices provides unique information regarding the peptide and protein expression in experimental models of PD. For example, dopamine depleted mice showed an increase in the expression of cytochrome C and cytochrome C oxidase, and that increased expression is most intense in the cortex. The present study demonstrates that protein profiles obtained from specific brain regions using DIGE and MALDI imaging combined with data from a peptidomic approach allow for the study of complex biochemical processes such as those occurring in experimental PD.
抄録全体を表示
-
南野 直人, 桑原 大幹, 松井 泰子, 磯山ー田中 純子, 木原 孝洋, 松八重 雅美, 高尾 敏文, 磯山 正治
セッションID: 2S1-4
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Peptidome database is aimed to accumulate the fact data for endogenous peptides based on their physicochemical properties including hydrophobicity, net charge and molecular weight. To construct this database, the crude peptide fraction was first subjected to gel filtration. Two peptide fractions of Mr<3,000 and 3,000<Mr<6,000 were each separated into 5,000 fractions by 2-dimensional (2D) HPLC (1st strong cation exchange (SCX) and 2nd reverse phase (RP) HPLC), and an aliquot of each fraction was analyzed with MALDI-TOF, ESI-Q-TOF and MALDI-TOF-TOF mass spectrometers (MSs). Starting from 1-2 g of tissue, we detected 16,788 peptides of Mr<6,000 in the pig brain and 4,058 peptides of Mr<3,000 in the mouse brain, and 26% of the peptides were identified with the tandem MSs. Among the identified peptides, about 20% were derived from peptide precursor proteins or secretory proteins, but about 30% were considered to be generated by degradation of the proteins. The biological activity of each fraction on the 2D-HPLC is also being evaluated and stored in the database. By combining these data, Peptidome database can be used as the powerful and intellectual basement for the discovery of new peptides.
We are also developing the efficient and quick peptide information acquisition system, which consists of automated 2D-HPLC (step-wise/linear and linear/linear) coupled on-line with ESI-TOF MS and MALDI-TOF-TOF MS. Three-fourths of the eluates from the 2D-HPLC were infused into the ESI-TOF MS, and the remaining portion was spotted on the target plates for MALDI-TOF/TOF MS. In the preliminary analysis, about 800 peptides and 160 peptides were detected and identified as for the pig brain peptides of Mr<3,000 obtained from 1.5 g of tissue.
抄録全体を表示
-
Eric Huang
セッションID: 2S2-1
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Our research emphasis is to develop noninvasive needle-free vaccines primarily based on the establishment of proteome profiles of various biological samples in our laboratory (J. Invest. Dermatol., 121: 51-64, 2003). Our platform is to develop epicutaneous and intranasal vaccines via adenoviral (DNA)-based and E. coil (protein)-based vectors. In this talk, we will take Bacillus anthracis as an example, highlight for you how we utilize proteomics to 1) reveal the mechanism of noninvasive vaccines and 2) find novel antigens from Bacillus anthracis. Furthermore, the core technique of noninvasive vaccines based on proteome profiles will be discussed. Theoretically, bioterrorists with bioengineering capabilities may expose a nation of people to anthrax spores producing toxins other than PA-LF-EF. We have identified and purified several germination-associated proteins from Bacillus anthracis by using proteomics. These proteins were differentially expressed from spore dormancy to germination. Eleven identified proteins have been characterized including one secreted protein (Immune inhibitor A), four potential membrane-associated proteins (Camelysin, Germination protease, Alanine racemase, and L-arabinose transporter), two molecular chaperones (HSP60 and cpn60), two energy-related proteins (ATP synthase and GPDH) and one novel anthrax specific protein. Our data demonstrated that Camelysin is a secretory toxin with cell selectivity. The noninvasive vaccines generated in our lab targeting Camelysin illustrated that Camelysin is an excellent antigen and provides protection from anthrax spore challenge. Needle-free epicutaneous or intranasal vaccinations are easily performed with minimal assistance from professional medical personnel during a bioterrorist attack or infectious disease epidemic. The ease of this technology is also beneficial for populations where medical personnel are lacking. Another purpose for introducing the noninvasive vaccine techniques in this talk is to seek diversified collaboration to develop skin patch vaccines targeting human infectious diseases such as Severe Acute Respiratory Syndrome (SARS).
抄録全体を表示
-
小田 吉哉
セッションID: 2S2-2
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Looking back over the past decade one has the impression of rapid biological breakthroughs but of chemical design knowledge advancing at a slower pace. Therefore compound-oriented approach is really needed for drug discovery.
Current commercial available drugs are mainly synthetic small compounds, and at least in near future, still such small molecules will be dominated on the drug market. Some of these compounds cause specific phenotypes (drug effects) to cells/animals, although these mode-of-actions are unknown. Chemical proteomic process uses synthetic small compounds to make clear their mechanism or to study protein functions. In pharmaceutical industries, it is very important to generate lead compound-drug target pairs with pharmacological significance. There are two types of chemical proteomics. One is the protein expression-based profiling of synthetic compounds to discover marker proteins and to identify the pathway of drug actions. Another is the drug target identification by using proteome technology.
Recent developments of quantitative proteome analysis using stable isotopes have rendered the protein-expression profiling in human cell lines reliable and feasible to look into a drug response pathway. We have developed new software to make possible the automated quantitation and annotation from large scale- data.
For primary target elucidation, synthetic small compounds are covalently immobilized to solid-phase as affinity matrix and subsequently allow proteins identification as drug targets. But compared with natural products, the specificity of synthetic compounds to their protein targets is rather low in many cases. Thus, I will talk about the quantitative manner to discriminate specific binders from nonspecific interactions.
抄録全体を表示
-
和田 忠士, 半田 宏
セッションID: 2S2-3
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Affinity purification is an established technique used to identify ligand-binding proteins, however, its widespread use has been limited by the inefficiency and instability of conventional matrices. The unstable nature of conventional matrices narrows the spectrum of ligands that can be used. Moreover, nonspecific binding of proteins to the solid support has complicated the identification of target proteins.
This study describes the preparation and use of affinity beads for the identification of drug receptors. The drugs of interest are immobilized onto a matrix that consists of latex beads covalently coupled with spacers. The latex beads are composed of glycidylmethacrylate (GMA)-covered GMA-styrene (St) copolymer cores (SG beads), which were originally developed for the affinity purification of DNA-binding proteins. To reduce steric hindrance, a divalent epoxide, ethyleneglycol diglycidylether (EGDE) molecules are introduced into the SG beads (SGE beads) as spacers after aminolysis of the epoxy groups on the surfaces of the beads.
The new SGE beads offer several distinct advantages compared to commonly used supports such as agarose beads. These advantages have enabled us to identify drug receptors directly from crude cell extracts within a few hours. Drug derivatives with a reactive group are covalently coupled to the SGE beads. A typical target protein is purified more than 1000 fold, with a yield of more than 70%. If necessary, the batch purification step can be repeated, further increasing the purity of the target protein. This procedure is not only effective, but is also simple and straightforward to perform. Using the affinity beads, we have identified multiple drugs receptors, which can then be used in such applications as drug screening, drug design, and evaluation of drug efficiency for given individuals.
抄録全体を表示
-
田中 明人, 原村 昌幸, 山崎 晃
セッションID: 2S2-4
発行日: 2004年
公開日: 2004/05/07
会議録・要旨集
フリー
Affinity chromatography matrices bearing bioactive compounds such as medicines, natural products, and toxins play an important role in the discovery of novel drug targets and the elucidation of drug mechanisms. Their effectiveness has been illustrated by finding of FKBP12, HDAC, Ref-13, and MDH as specific binding proteins of FK506 (1), Trapoxin, E-3330, and E-7070, respectively. The successful isolation of target proteins by affinity chromatography depends on the synthesis of polymeric resins that can bind to the cellular target with maximum selectivity and efficiency. Tubulin and actin often bind non-specifically to affinity chromatography resins, complicating research toward identifying the cellular targets of small molecules. Reduction of non-specific binding proteins is important for the success of such biochemical approaches. In order to develop strategies to circumvent this problem, we quantitatively investigated the binding of tubulin and actin to a series of affinity resins bearing fifteen variant ligands on three commercially available polymer supports. Non-specific protein binding was proportional to the hydrophobicity of the affinity resins and could be quantitatively correlated to the CLOGP values of the ligands, which are a measure of compound hydrophobicity. When compounds had CLOGP values greater than 1.5, (tubulin)=0.73*CLOGP-1.1, and (actin)=0.42*CLOGP-0.79 (n=7). Based on these studies, we designed a novel hydrophilic polyethyleneglycol (PEG) spacer for the conjugation of ligands to chromatography resins. As predicted by our binding algorithm, introduction of this spacer reduced the amount of non-specific protein binding in proportion to number of ethylene glycol units. If we have enough time, we additionally show a simple and versatile method for immobilization of ligands, termed "Chemical Shotgun", in which compounds are non-selectively immobilized on resins without the prior and boring design and synthesis of the derivative.
抄録全体を表示

