-
Hiroshi SAKIYAMA, Misaki ITO, Ryoji MITSUHASHI, Masahiro MIKURIYA
2016Volume 15Issue 3 Pages
49-50
Published: 2016
Released on J-STAGE: October 01, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
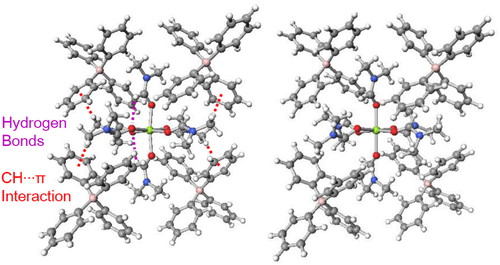
In a crystal, an octahedral magnesium(II) complex cation, [Mg(DMF)6]2+ (DMF: N,N-dimethylformamide), was found to exist as a C2 conformer, which was thought to be energetically unfavorable. In order to find the reason for this, structural analysis was conducted on the basis of semi-empirical PM6 method and density functional theory method. The C2 conformer was not found to be stable in a vacuum; however, it was found to be stabilized when surrounded by four tetraphenyl borate anions like the crystal structure.
View full abstract
-
Shutaro KAWADA, Masataka SAKAGUCHI, Ibuki YONEKURA, Kouji OKUWAKI, Yuj ...
2016Volume 15Issue 3 Pages
51-52
Published: 2016
Released on J-STAGE: October 01, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
Peptoids are a class of peptide mimetics whose side chains contain nitrogen atoms rather than α-carbon atoms. This structural feature restricts the intrinsic capacity to form hydrogen bond networks of α-helix, therefore peptoid oligomers are attracting attention due to the ability to design chemical properties such as self-assembling by selecting proper side chains. This paper shows some illustrative peptoid calculations, based on the fragment molecular orbital (FMO) method.
View full abstract
-
Naohiro NISHIDA, Yuka HORIKAWA, Takashi TOKUSHIMA, Osamu TAKAHASHI
2016Volume 15Issue 3 Pages
53-54
Published: 2016
Released on J-STAGE: October 01, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
We performed theoretically to reproduce site-selective X-ray emission spectroscopy (XES) spectra of carbonate in the liquid phase at the oxygen K-edge. Structure sampling as a cluster model was performed from a snapshot of the first principles molecular dynamics simulation. Relative intensities of XES with core-hole excited state dynamics simulation were calculated using density functional theory. Theoretical XES spectra for CO32- and HCO3− were well reproduced experimentally and that for H2CO3 was predicted.
View full abstract
-
Hideyuki NARUMI
2016Volume 15Issue 3 Pages
55-56
Published: 2016
Released on J-STAGE: October 01, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
We changed a mass formula obtained by the Heisenburg Method into one expressed in Schroedinger style and tried to clarify its physical meanings including of the origin of generations.
View full abstract
-
Dai MORIKAWA, Yasushi NOMURA, Noriyuki MIZOGUCHI
2016Volume 15Issue 3 Pages
57-59
Published: 2016
Released on J-STAGE: October 08, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
Graphyne nanotubes (GNTs) are new carbon-based nanotubes and have a structure similar to the carbon nanotubes (CNTs). In the GNTs, armchair α-GNT is the highest resemblance to armchair CNT, and algebraic structure counts (ASC) are equal. This correspondence suggests that the HOMO-LUMO gaps in the finite-length armchair α-GNT oscillate with the increase of the tube length in the same fashion as the oscillation of HOMO-LUMO gaps in finite-length armchair CNT. In this work, the tube length dependences of the HOMO-LUMO gaps in the finite-length armchair α-GNT were examined by means of the Hückel method and density functional theory (DFT). It was found that the HOMO-LUMO gaps in armchair α-GNT oscillate as functions of the tube length with the period of 3.
View full abstract
-
Makoto ISOGAI, Hirohiko HOUJOU
2016Volume 15Issue 3 Pages
60-62
Published: 2016
Released on J-STAGE: October 08, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
We have developed a method for quantifying the intermolecular forces in supramolecular systems by modifying our original method of coarse-graining intermolecular vibrations. We evaluated the true and apparent intermolecular stiffness constants for 21 dimers composed of formic acid-, acetic acid-, trichloroacetic acid-, formamide-, formamidine- or urea-monomer. In this method, the atomic displacement vectors of a dimer were projected onto a subspace spanned by bases corresponding to 12 relative translational and rotational motions and several intramolecular vibrations that are coupled with intermolecular vibrations. The intermolecular stiffness constants showed moderate linearity with the corresponding dimerization energies. The apparent stiffness constant can be explained by a mechanical model using inter- and intramolecular stiffness constants of the constituent monomers.
View full abstract
-
Mikito FUJINAMI, Junji SEINO, Hiromi NAKAI
2016Volume 15Issue 3 Pages
63-65
Published: 2016
Released on J-STAGE: October 08, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
We have developed a novel reaction prediction system, which uses machine learning with quantum chemical descriptors. Numerical assessments of the system were performed on basic polar and radical organic chemical reactions. The accuracy of the present system was close to that of a previous system having machine learning with topological information, which is termed ReactionPredictor.
View full abstract
-
Yuji MOCHIZUKI, Shota NAKAMURA, Masahiro YAMANAKA, Yasuyuki YAMADA, Mi ...
2016Volume 15Issue 3 Pages
66-67
Published: 2016
Released on J-STAGE: October 08, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
Recently, technologies and applications of 3D-printers have attracted practical interests in the contexts of manufacturing and research developments. In contrast, the educational usages have still been underway. In this Letter, we report a variety of demonstrative 3D-printed molecular models used for education of chemistry and biology in our faculty of Science.
View full abstract
-
Yuya NAKAJIMA, Junji SEINO, Michael W. SCHMIDT, Hiromi NAKAI
2016Volume 15Issue 3 Pages
68-70
Published: 2016
Released on J-STAGE: October 08, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
This Letter provides an implementation of an efficient and accurate relativistic method based on the infinite-order two-component scheme with the local unitary transformation (LUT-IOTC) to the GAMESS program. The sample input and major capabilities in GAMESS are shown as well as the accuracies and efficiencies in energy and analytical energy gradient calculations. The scheme realizes calculations of molecules containing heavy elements with four-component relativistic accuracy and the non-relativistic computational cost.
View full abstract
-
Yoshiaki TANIDA, Azuma MATSUURA
2016Volume 15Issue 3 Pages
71-73
Published: 2016
Released on J-STAGE: October 19, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
We have performed metadynamics to explore the ligand docking poses with an RNA aptamer in three collective variables (CVs) space, one distance and two dihedral angles. We showed that the free energy surface (FES) of the ligand binding has several local minima. Furthermore, we also demonstrate that each metastable structure can be deduced from CVs of each free energy minimum.
View full abstract
-
Hiroyuki SATO, Azuma MATSUURA
2016Volume 15Issue 3 Pages
74-76
Published: 2016
Released on J-STAGE: October 19, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
The hydration structure in the active site of human coagulation factor Xa (fXa) was investigated from the viewpoint of excess free energy. Water distribution in the active site of fXa was calculated using partly constrained molecular dynamics (MD) simulation. Then the free energy of the distribution was evaluated using grid inhomogeneous solvation theory (GIST). The analyzed excess free energy shows excellent correlation to the experimental binding affinity of known ligands only if the side chain fluctuation of the active site of fXa was taken into account. This result indicates that the side chain fluctuation generated by partly constrained MD has an important role for identifying the appropriate hydration structure, and that GIST in combination with partly constrained MD provides useful information for assessing the protein-ligand binding affinity.
View full abstract
-
Shohei KANNO, Yutaka IMAMURA, Masahiko HADA
2016Volume 15Issue 3 Pages
77-78
Published: 2016
Released on J-STAGE: November 04, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
We theoretically examine spin-forbidden excitations for an N3-skeleton Ru dye with a thienyl group and iodine anion by time-dependent density functional theory with spin-orbit coupling. We found that the oscillator strength in the near-infrared region and spin-orbit coupling are enhanced by introducing the thienyl group into bipirydyl ligands.
View full abstract
-
Ryoji FUJIYAMA, Risako HIDAKA
2016Volume 15Issue 3 Pages
79-80
Published: 2016
Released on J-STAGE: November 04, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
Substituent effects in cross-conjugated systems were carried out on a series of substituted 3-methylenepenta- 1,4-diynes with primary carbocation CH2+. Their optimized energies were calculated at the B3LYP/6-31G (d) level. The substituent effects were expressed in terms of isodesmic reactions and were analyzed by LSFE equation. The analysis results show that the substituent effect is inversely proportional to the stability of the whole molecule. In other words, the more stable the cross-conjugated molecule is, the smaller it's substituent effect become.
View full abstract
-
Kaoruho SAKATA, Takashi HISATOMI, Yosuke GOTO, Blanka MAGYARI-KÖPE, Pe ...
2016Volume 15Issue 3 Pages
81-82
Published: 2016
Released on J-STAGE: November 05, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
La5Ti2Cu1-xAgxS5O7, the solid solution of La5Ti2CuS5O7 and La5Ti2AgS5O7, is a candidate of photocatalysts for sunlight-driven water splitting. It is known that La5Ti2Cu0.9Ag0.1S5O7 has a narrower band gap than La5Ti2CuS5O7 and La5Ti2AgS5O7. In this study, density functional theory (DFT) calculation was performed to analyze the band structure and effective mass of La5Ti2Cu1-xAgxS5O7 solid solutions. The band gap of La5Ti2Cu0.75Ag0.25S5O7 was calculated to be narrower than those of La5Ti2CuS5O7 and La5Ti2AgS5O7, which was qualitatively consistent with the experimental observation. The effective mass for electron was also estimated from the band structure to be smaller in the b-axis direction than in the other directions.
View full abstract
-
Shinsaku FUJITA
2016Volume 15Issue 3 Pages
83-84
Published: 2016
Released on J-STAGE: November 18, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
An interdisciplinary book entitled “Mathematical Stereochemistry” has been published by means of the combination of freely-available computer systems, i.e., the LATEX system for typesetting mathematical formulas and the XyMTEX system for drawing chemical structural formulas.
View full abstract
-
Takayoshi ISHIMOTO, Michihisa KOYAMA
2016Volume 15Issue 3 Pages
85-86
Published: 2016
Released on J-STAGE: November 18, 2016
JOURNAL
FREE ACCESS
FULL-TEXT HTML
We analyzed the structural differences of H2O molecules in water nanodroplet on graphene ((H2O)98 on graphene) model by using the density functional theory. 98 H2O molecules on graphene were classified into four groups based on the surrounding condition (bulk region, water-gas interface, water-graphene interface, and water-graphene-gas interface). The O–H distances and vibrational frequencies of H2O molecules near the gas region were wider distributions compared with the H2O molecules in the bulk region, whereas narrower distributions were obtained near the graphene interface.
View full abstract

 Download PDF (952K) Full view HTML
Download PDF (952K) Full view HTML Download PDF (1061K) Full view HTML
Download PDF (1061K) Full view HTML Download PDF (964K) Full view HTML
Download PDF (964K) Full view HTML Download PDF (838K) Full view HTML
Download PDF (838K) Full view HTML