
89 巻, 2 号
選択された号の論文の27件中1~27を表示しています
- |<
- <
- 1
- >
- >|
The 65th special feature “Fluorine Chemistry and Materials for Electrochemistry”
Comprehensive Paper
-
原稿種別: The 65th special feature “Fluorine Chemistry and Materials for Electrochemistry”
2021 年 89 巻 2 号 p. 75-82
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/12/11PDF形式でダウンロード (1122K)
Communication
-
原稿種別: The 65th special feature “Fluorine Chemistry and Materials for Electrochemistry”
2021 年 89 巻 2 号 p. 83-86
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/12/16 PDF形式でダウンロード (2464K)
PDF形式でダウンロード (2464K)
Articles
-
原稿種別: The 65th special feature “Fluorine Chemistry and Materials for Electrochemistry”
2021 年 89 巻 2 号 p. 87-93
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/11/12PDF形式でダウンロード (2597K) -
原稿種別: The 65th special feature “Fluorine Chemistry and Materials for Electrochemistry”
2021 年 89 巻 2 号 p. 94-99
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/11/28PDF形式でダウンロード (4202K) -
原稿種別: The 65th special feature “Fluorine Chemistry and Materials for Electrochemistry”
2021 年 89 巻 2 号 p. 100-103
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/12/17 PDF形式でダウンロード (1816K)
PDF形式でダウンロード (1816K) -
原稿種別: The 65th special feature “Fluorine Chemistry and Materials for Electrochemistry”
2021 年 89 巻 2 号 p. 104-110
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/13PDF形式でダウンロード (2317K) -
原稿種別: The 65th special feature “Fluorine Chemistry and Materials for Electrochemistry”
2021 年 89 巻 2 号 p. 111-117
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/26 PDF形式でダウンロード (2013K)
PDF形式でダウンロード (2013K)
Notes
-
原稿種別: The 65th special feature “Fluorine Chemistry and Materials for Electrochemistry”
2021 年 89 巻 2 号 p. 118-120
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/12/10PDF形式でダウンロード (810K) -
原稿種別: The 65th special feature “Fluorine Chemistry and Materials for Electrochemistry”
2021 年 89 巻 2 号 p. 121-124
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/13PDF形式でダウンロード (1534K) -
原稿種別: The 65th special feature “Fluorine Chemistry and Materials for Electrochemistry”
2021 年 89 巻 2 号 p. 125-130
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/13 PDF形式でダウンロード (1559K)
PDF形式でダウンロード (1559K)
Regular Papers
Communications
-
2021 年 89 巻 2 号 p. 131-133
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/11/19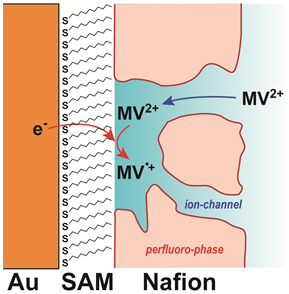 PDF形式でダウンロード (1478K)
PDF形式でダウンロード (1478K) -
2021 年 89 巻 2 号 p. 134-137
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/12/04PDF形式でダウンロード (1513K) -
2021 年 89 巻 2 号 p. 138-140
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/12/05 PDF形式でダウンロード (842K)
PDF形式でダウンロード (842K) -
2021 年 89 巻 2 号 p. 141-144
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/13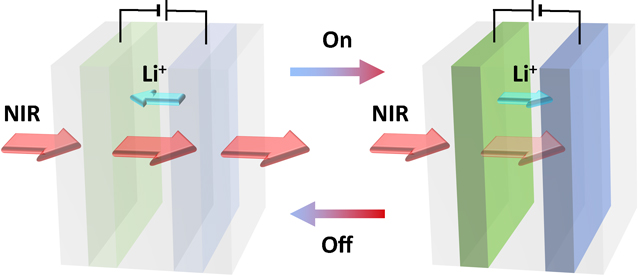 PDF形式でダウンロード (1849K)
PDF形式でダウンロード (1849K) -
2021 年 89 巻 2 号 p. 145-147
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/28PDF形式でダウンロード (1481K)
Articles
-
2021 年 89 巻 2 号 p. 148-156
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/11/21 PDF形式でダウンロード (4575K)
PDF形式でダウンロード (4575K) -
2021 年 89 巻 2 号 p. 157-161
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/11/25PDF形式でダウンロード (3598K) -
2021 年 89 巻 2 号 p. 162-166
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/12/08PDF形式でダウンロード (2769K) -
2021 年 89 巻 2 号 p. 167-175
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/12/12PDF形式でダウンロード (10526K) -
2021 年 89 巻 2 号 p. 176-185
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2020/12/26PDF形式でダウンロード (6286K) -
2021 年 89 巻 2 号 p. 186-191
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/09PDF形式でダウンロード (1648K) -
2021 年 89 巻 2 号 p. 192-196
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/19PDF形式でダウンロード (3220K) -
2021 年 89 巻 2 号 p. 197-203
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/23 PDF形式でダウンロード (1644K)
PDF形式でダウンロード (1644K) -
2021 年 89 巻 2 号 p. 204-210
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/23 PDF形式でダウンロード (3999K)
PDF形式でダウンロード (3999K) -
2021 年 89 巻 2 号 p. 211-214
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/29PDF形式でダウンロード (2893K)
Notes
-
2021 年 89 巻 2 号 p. 215-217
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/09 PDF形式でダウンロード (1219K)
PDF形式でダウンロード (1219K) -
2021 年 89 巻 2 号 p. 218-222
発行日: 2021/03/05
公開日: 2021/03/05
[早期公開] 公開日: 2021/01/26 PDF形式でダウンロード (3757K)
PDF形式でダウンロード (3757K)
- |<
- <
- 1
- >
- >|