Volume 61, Issue 1
Displaying 1-31 of 31 articles from this issue
- |<
- <
- 1
- >
- >|
Review
-
Effect of Alloying Elements on Fracture Toughness and Ductility in Magnesium Binary Alloys; A ReviewArticle type: Review
2020 Volume 61 Issue 1 Pages 1-13
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: September 30, 2019 Download PDF (6352K) Full view HTML
Download PDF (6352K) Full view HTML -
Article type: Review
2020 Volume 61 Issue 1 Pages 14-26
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
 Download PDF (8751K) Full view HTML
Download PDF (8751K) Full view HTML
Special Issue on New Aspects of Martensitic Transformations II
-
Article type: Regular Article
Subject area: Special Issue on New Aspects of Martensitic Transformations II
2020 Volume 61 Issue 1 Pages 27-32
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: September 24, 2019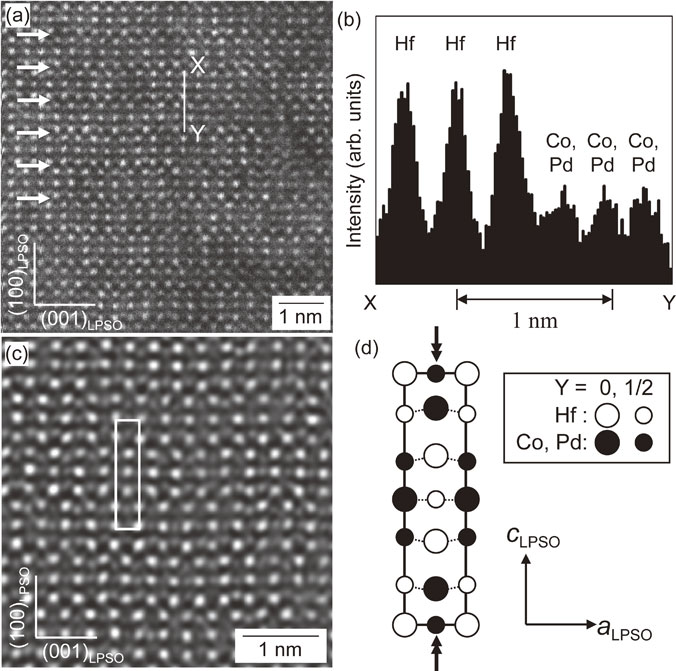 Download PDF (3815K) Full view HTML
Download PDF (3815K) Full view HTML -
Article type: Regular Article
Subject area: Special Issue on New Aspects of Martensitic Transformations II
2020 Volume 61 Issue 1 Pages 33-36
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: September 24, 2019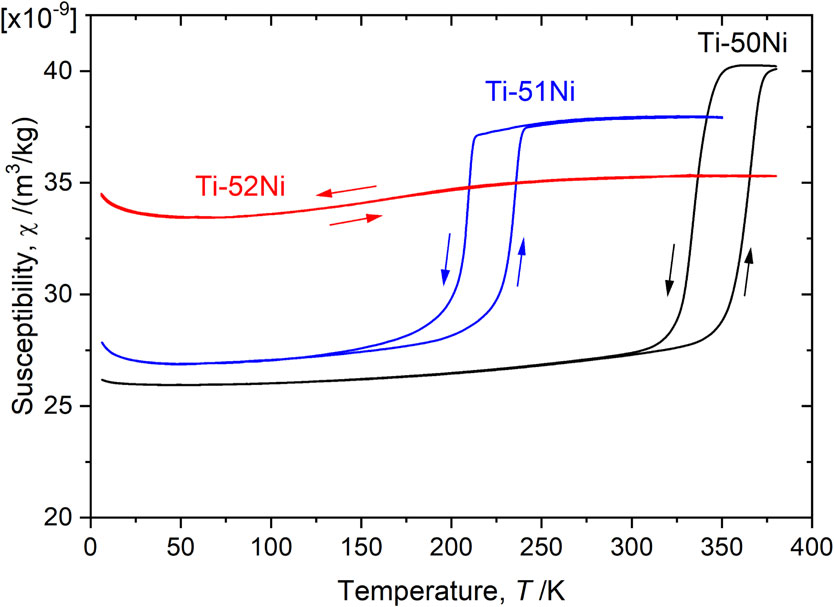 Download PDF (1273K) Full view HTML
Download PDF (1273K) Full view HTML -
Article type: Regular Article
Subject area: Special Issue on New Aspects of Martensitic Transformations II
2020 Volume 61 Issue 1 Pages 37-41
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: September 30, 2019 Download PDF (3388K) Full view HTML
Download PDF (3388K) Full view HTML -
Article type: Regular Article
Subject area: Special Issue on New Aspects of Martensitic Transformations II
2020 Volume 61 Issue 1 Pages 42-48
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: October 04, 2019 Download PDF (2215K) Full view HTML
Download PDF (2215K) Full view HTML -
Article type: Regular Article
Subject area: Special Issue on New Aspects of Martensitic Transformations II
2020 Volume 61 Issue 1 Pages 49-54
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
 Download PDF (3647K) Full view HTML
Download PDF (3647K) Full view HTML -
Article type: Regular Article
Subject area: Special Issue on New Aspects of Martensitic Transformations II
2020 Volume 61 Issue 1 Pages 55-60
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: October 21, 2019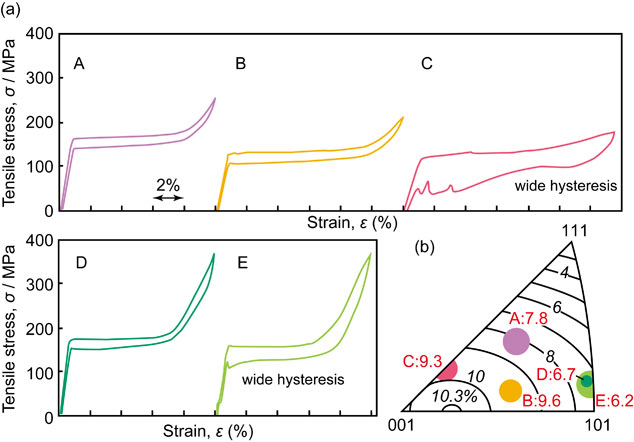 Download PDF (2515K) Full view HTML
Download PDF (2515K) Full view HTML -
Article type: Regular Article
Subject area: Special Issue on New Aspects of Martensitic Transformations II
2020 Volume 61 Issue 1 Pages 61-67
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
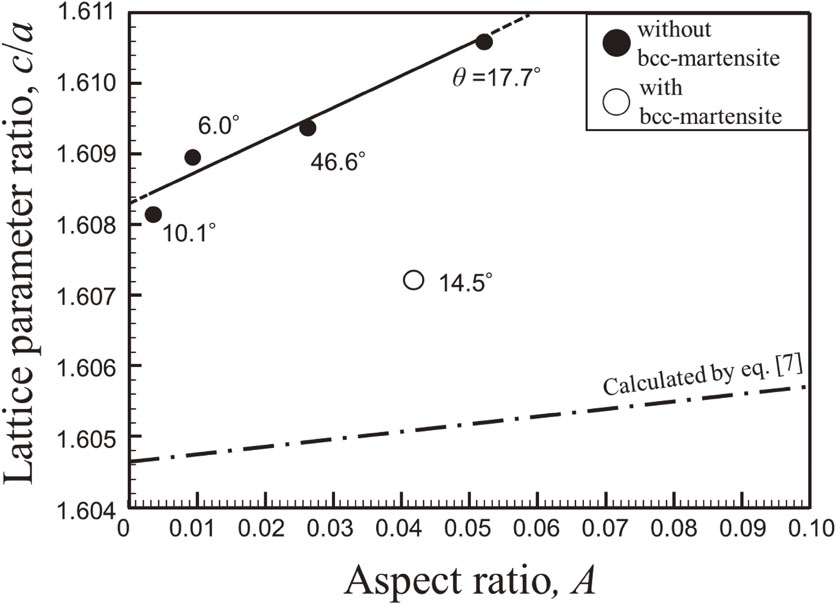 Download PDF (3802K) Full view HTML
Download PDF (3802K) Full view HTML -
Article type: Express Rapid Publication
Subject area: Special Issue on New Aspects of Martensitic Transformations Ⅱ
2020 Volume 61 Issue 1 Pages 68-71
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 15, 2019 Download PDF (2032K) Full view HTML
Download PDF (2032K) Full view HTML
Regular Article
Materials Physics
-
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 72-77
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 22, 2019 Download PDF (3072K) Full view HTML
Download PDF (3072K) Full view HTML -
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 78-87
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 22, 2019 Download PDF (2696K) Full view HTML
Download PDF (2696K) Full view HTML -
Atomic Layer Deposition of AlGaN on GaN and Current Transport Mechanism in AlGaN/GaN Schottky DiodesArticle type: Regular Article
2020 Volume 61 Issue 1 Pages 88-93
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 15, 2019 Download PDF (2371K) Full view HTML
Download PDF (2371K) Full view HTML -
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 94-103
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
 Download PDF (3016K) Full view HTML
Download PDF (3016K) Full view HTML
Microstructure of Materials
-
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 104-110
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 15, 2019 Download PDF (5380K) Full view HTML
Download PDF (5380K) Full view HTML
Mechanics of Materials
-
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 111-118
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 11, 2019 Download PDF (5447K) Full view HTML
Download PDF (5447K) Full view HTML -
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 119-126
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
 Download PDF (7278K) Full view HTML
Download PDF (7278K) Full view HTML -
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 127-135
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
 Download PDF (5912K) Full view HTML
Download PDF (5912K) Full view HTML
Materials Chemistry
-
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 136-141
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 22, 2019 Download PDF (1665K) Full view HTML
Download PDF (1665K) Full view HTML -
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 142-149
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
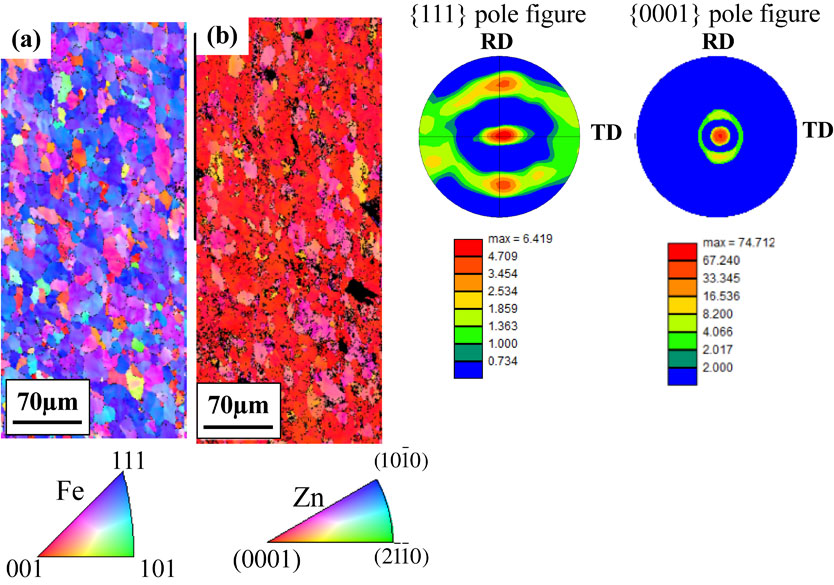 Download PDF (11348K) Full view HTML
Download PDF (11348K) Full view HTML -
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 150-155
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 22, 2019 Download PDF (929K) Full view HTML
Download PDF (929K) Full view HTML
Materials Processing
-
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 156-161
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 11, 2019Download PDF (2961K) Full view HTML -
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 162-168
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Download PDF (2694K) Full view HTML -
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 169-175
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 22, 2019Download PDF (4240K) Full view HTML -
Mechanical Properties Prediction of Gray Cast Iron Considering Trace Elements Based on Deep LearningArticle type: Regular Article
2020 Volume 61 Issue 1 Pages 176-180
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 22, 2019Download PDF (2643K) Full view HTML -
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 181-187
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
 Download PDF (5440K) Full view HTML
Download PDF (5440K) Full view HTML
Engineering Materials and Their Applications
-
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 188-194
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 11, 2019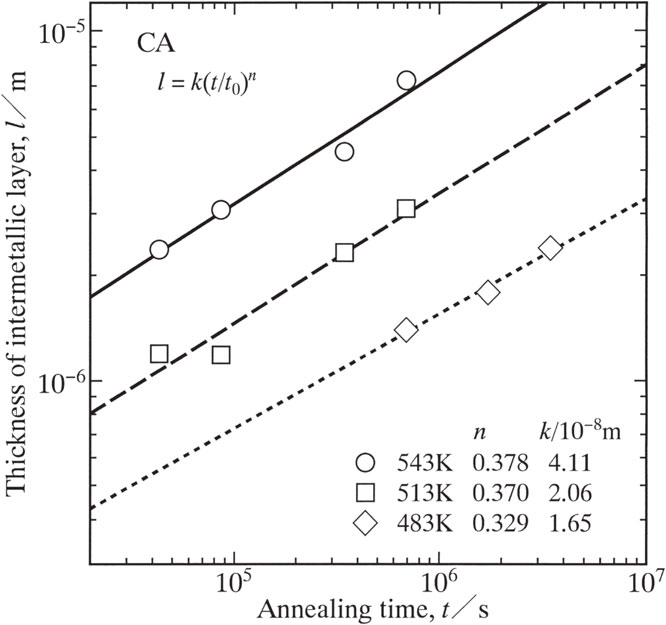 Download PDF (2674K) Full view HTML
Download PDF (2674K) Full view HTML -
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 195-199
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 11, 2019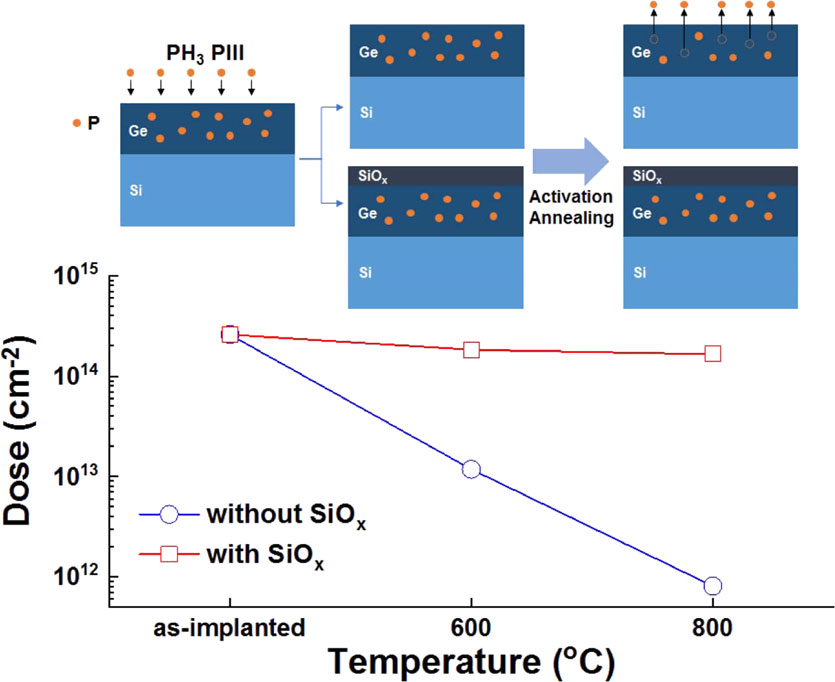 Download PDF (2267K) Full view HTML
Download PDF (2267K) Full view HTML -
Performance of AZ31 Alloy as Anodes for Primary Magnesium-Air Batteries under High Current DischargeArticle type: Regular Article
2020 Volume 61 Issue 1 Pages 200-205
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
 Download PDF (3548K) Full view HTML
Download PDF (3548K) Full view HTML -
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 206-212
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 29, 2019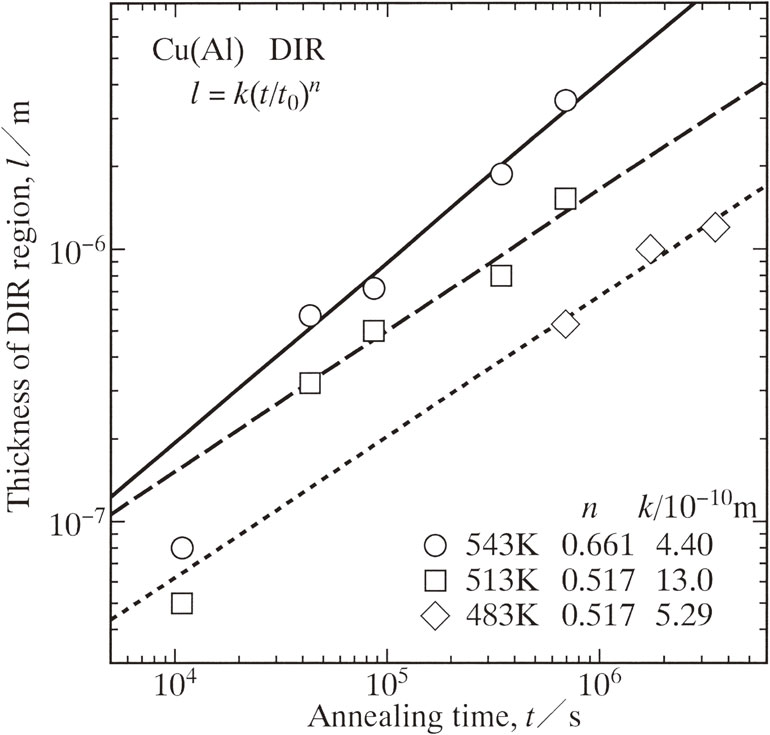 Download PDF (2284K) Full view HTML
Download PDF (2284K) Full view HTML -
Article type: Regular Article
2020 Volume 61 Issue 1 Pages 213-220
Published: January 01, 2020
Released on J-STAGE: December 25, 2019
Advance online publication: November 29, 2019 Download PDF (3251K) Full view HTML
Download PDF (3251K) Full view HTML
- |<
- <
- 1
- >
- >|