
17 巻, 3 号
日本コンピュータ化学会2018春季年会精選論文特集号
選択された号の論文の19件中1~19を表示しています
- |<
- <
- 1
- >
- >|
巻頭言
-
2018 年 17 巻 3 号 p. A15
発行日: 2018年
公開日: 2018/10/06
PDF形式でダウンロード (316K) HTML形式で全画面表示
速報
-
2018 年 17 巻 3 号 p. 111-112
発行日: 2018年
公開日: 2018/10/06
 PDF形式でダウンロード (1863K) HTML形式で全画面表示
PDF形式でダウンロード (1863K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 113-116
発行日: 2018年
公開日: 2018/10/06
 PDF形式でダウンロード (933K) HTML形式で全画面表示
PDF形式でダウンロード (933K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 117-119
発行日: 2018年
公開日: 2018/10/10
 PDF形式でダウンロード (768K) HTML形式で全画面表示
PDF形式でダウンロード (768K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 120-121
発行日: 2018年
公開日: 2018/10/16
PDF形式でダウンロード (497K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 122-123
発行日: 2018年
公開日: 2018/10/16
 PDF形式でダウンロード (573K) HTML形式で全画面表示
PDF形式でダウンロード (573K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 124-126
発行日: 2018年
公開日: 2018/10/16
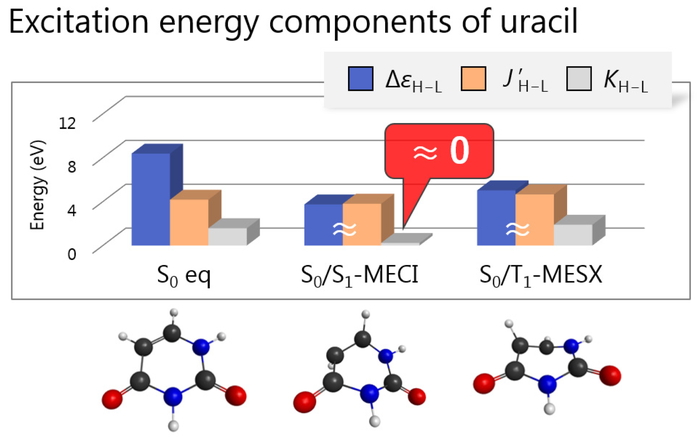 PDF形式でダウンロード (1108K) HTML形式で全画面表示
PDF形式でダウンロード (1108K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 127-129
発行日: 2018年
公開日: 2018/10/16
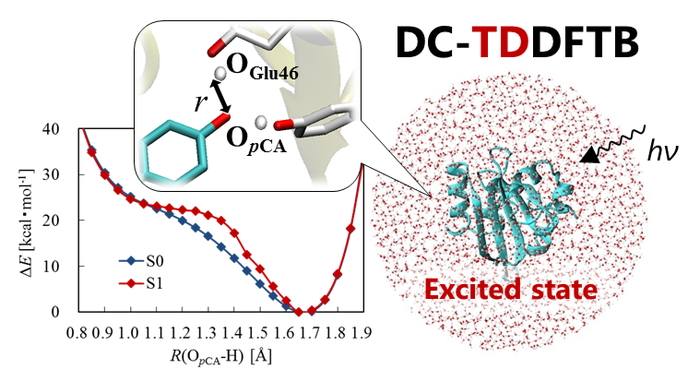 PDF形式でダウンロード (964K) HTML形式で全画面表示
PDF形式でダウンロード (964K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 130-132
発行日: 2018年
公開日: 2018/10/16
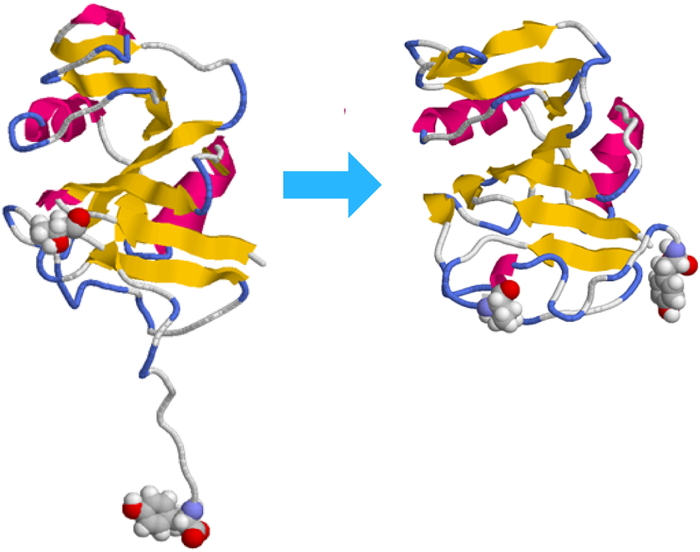 PDF形式でダウンロード (960K) HTML形式で全画面表示
PDF形式でダウンロード (960K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 133-137
発行日: 2018年
公開日: 2018/10/16
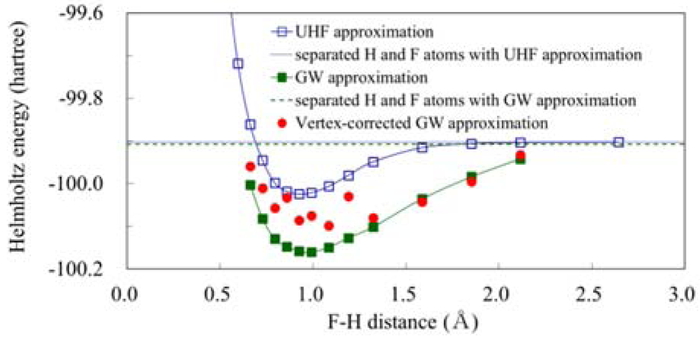 PDF形式でダウンロード (740K) HTML形式で全画面表示
PDF形式でダウンロード (740K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 138-141
発行日: 2018年
公開日: 2018/10/22
 PDF形式でダウンロード (1573K) HTML形式で全画面表示
PDF形式でダウンロード (1573K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 142-143
発行日: 2018年
公開日: 2018/10/26
 PDF形式でダウンロード (495K) HTML形式で全画面表示
PDF形式でダウンロード (495K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 144-146
発行日: 2018年
公開日: 2018/10/26
 PDF形式でダウンロード (1372K) HTML形式で全画面表示
PDF形式でダウンロード (1372K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 147-149
発行日: 2018年
公開日: 2018/11/02
 PDF形式でダウンロード (1037K) HTML形式で全画面表示
PDF形式でダウンロード (1037K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 150-152
発行日: 2018年
公開日: 2018/11/10
 PDF形式でダウンロード (561K) HTML形式で全画面表示
PDF形式でダウンロード (561K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 153-154
発行日: 2018年
公開日: 2018/11/10
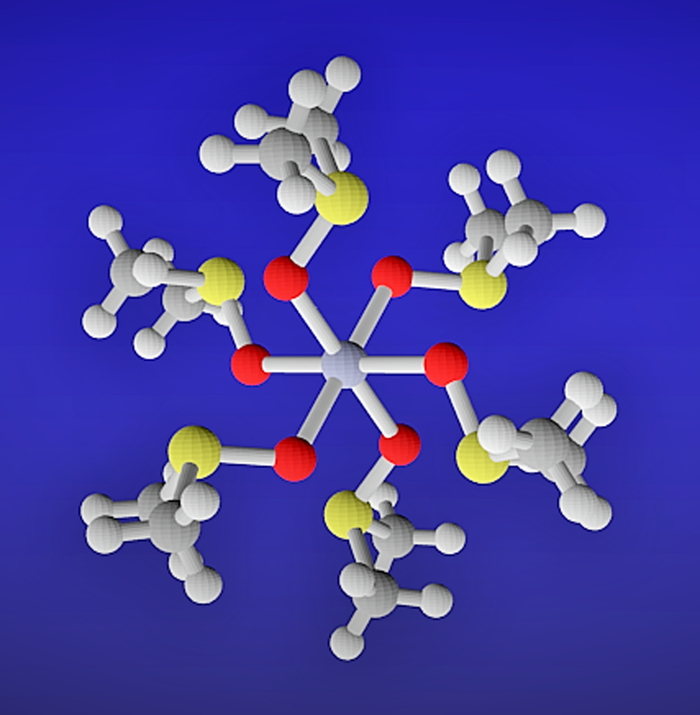 PDF形式でダウンロード (689K) HTML形式で全画面表示
PDF形式でダウンロード (689K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 155-157
発行日: 2018年
公開日: 2018/11/10
 PDF形式でダウンロード (1282K) HTML形式で全画面表示
PDF形式でダウンロード (1282K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 158-159
発行日: 2018年
公開日: 2018/11/17
PDF形式でダウンロード (1523K) HTML形式で全画面表示 -
2018 年 17 巻 3 号 p. 160-162
発行日: 2018年
公開日: 2018/12/21
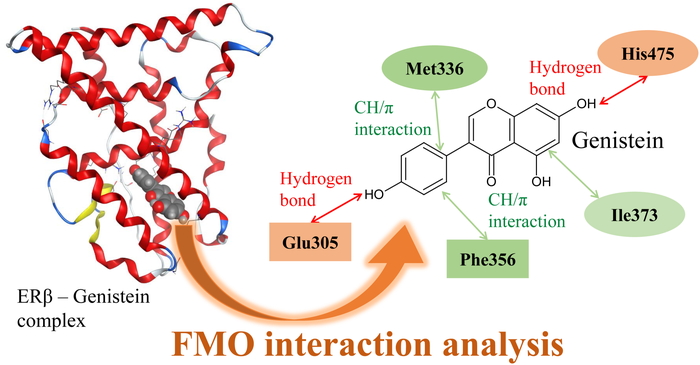 PDF形式でダウンロード (1044K) HTML形式で全画面表示
PDF形式でダウンロード (1044K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|