
15 巻, 3 号
日本コンピュータ化学会2016 春季年会精選論文特集号
選択された号の論文の17件中1~17を表示しています
- |<
- <
- 1
- >
- >|
巻頭言
-
2016 年 15 巻 3 号 p. A39
発行日: 2016年
公開日: 2016/10/01
PDF形式でダウンロード (336K) HTML形式で全画面表示
速報
-
2016 年 15 巻 3 号 p. 49-50
発行日: 2016年
公開日: 2016/10/01
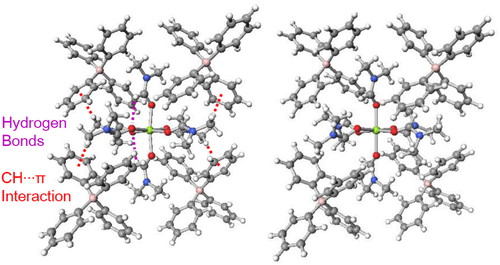 PDF形式でダウンロード (952K) HTML形式で全画面表示
PDF形式でダウンロード (952K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 51-52
発行日: 2016年
公開日: 2016/10/01
PDF形式でダウンロード (891K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 53-54
発行日: 2016年
公開日: 2016/10/01
 PDF形式でダウンロード (1061K) HTML形式で全画面表示
PDF形式でダウンロード (1061K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 55-56
発行日: 2016年
公開日: 2016/10/01
PDF形式でダウンロード (413K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 57-59
発行日: 2016年
公開日: 2016/10/08
PDF形式でダウンロード (758K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 60-62
発行日: 2016年
公開日: 2016/10/08
 PDF形式でダウンロード (964K) HTML形式で全画面表示
PDF形式でダウンロード (964K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 63-65
発行日: 2016年
公開日: 2016/10/08
 PDF形式でダウンロード (838K) HTML形式で全画面表示
PDF形式でダウンロード (838K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 66-67
発行日: 2016年
公開日: 2016/10/08
PDF形式でダウンロード (846K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 68-70
発行日: 2016年
公開日: 2016/10/08
PDF形式でダウンロード (660K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 71-73
発行日: 2016年
公開日: 2016/10/19
PDF形式でダウンロード (1340K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 74-76
発行日: 2016年
公開日: 2016/10/19
PDF形式でダウンロード (973K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 77-78
発行日: 2016年
公開日: 2016/11/04
PDF形式でダウンロード (818K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 79-80
発行日: 2016年
公開日: 2016/11/04
PDF形式でダウンロード (552K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 81-82
発行日: 2016年
公開日: 2016/11/05
PDF形式でダウンロード (601K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 83-84
発行日: 2016年
公開日: 2016/11/18
PDF形式でダウンロード (515K) HTML形式で全画面表示 -
2016 年 15 巻 3 号 p. 85-86
発行日: 2016年
公開日: 2016/11/18
PDF形式でダウンロード (516K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|