
39 巻, 5 号
選択された号の論文の33件中1~33を表示しています
- |<
- <
- 1
- >
- >|
Regular Articles
-
2016 年 39 巻 5 号 p. 653-656
発行日: 2016/05/01
公開日: 2016/05/01
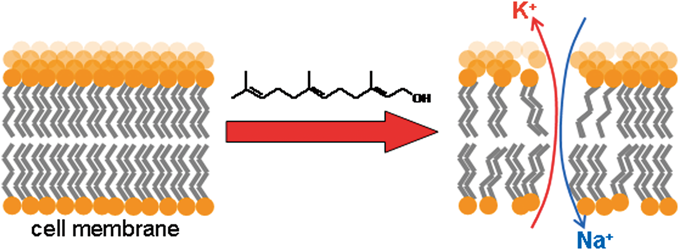 PDF形式でダウンロード (572K) HTML形式で全画面表示
PDF形式でダウンロード (572K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 657-664
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (1335K) HTML形式で全画面表示
PDF形式でダウンロード (1335K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 665-673
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (8252K) HTML形式で全画面表示
PDF形式でダウンロード (8252K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 674-679
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/02/25 PDF形式でダウンロード (441K) HTML形式で全画面表示
PDF形式でダウンロード (441K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 680-688
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/03/05 PDF形式でダウンロード (2380K) HTML形式で全画面表示
PDF形式でダウンロード (2380K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 689-698
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (989K) HTML形式で全画面表示
PDF形式でダウンロード (989K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 699-704
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/02/18 PDF形式でダウンロード (433K) HTML形式で全画面表示
PDF形式でダウンロード (433K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 705-711
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (773K) HTML形式で全画面表示
PDF形式でダウンロード (773K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 712-720
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (4952K) HTML形式で全画面表示
PDF形式でダウンロード (4952K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 721-727
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/02/26 PDF形式でダウンロード (382K) HTML形式で全画面表示
PDF形式でダウンロード (382K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 728-736
発行日: 2016/05/01
公開日: 2016/05/01
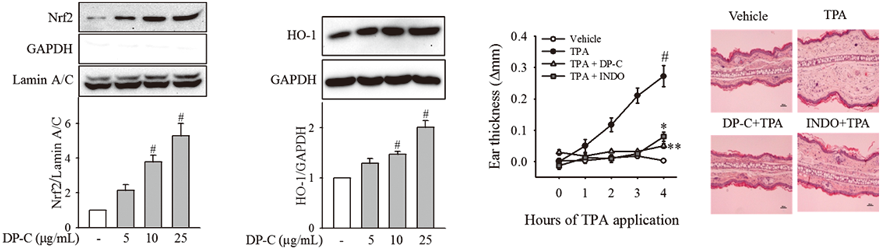 PDF形式でダウンロード (1873K) HTML形式で全画面表示
PDF形式でダウンロード (1873K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 737-746
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/02/22 PDF形式でダウンロード (7731K) HTML形式で全画面表示
PDF形式でダウンロード (7731K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 747-753
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/03/01 PDF形式でダウンロード (1144K) HTML形式で全画面表示
PDF形式でダウンロード (1144K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 754-761
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (738K) HTML形式で全画面表示
PDF形式でダウンロード (738K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 762-769
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/03/03 PDF形式でダウンロード (1824K) HTML形式で全画面表示
PDF形式でダウンロード (1824K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 770-777
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (4394K) HTML形式で全画面表示
PDF形式でダウンロード (4394K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 778-785
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (3080K) HTML形式で全画面表示
PDF形式でダウンロード (3080K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 786-793
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (1251K) HTML形式で全画面表示
PDF形式でダウンロード (1251K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 794-798
発行日: 2016/05/01
公開日: 2016/05/01
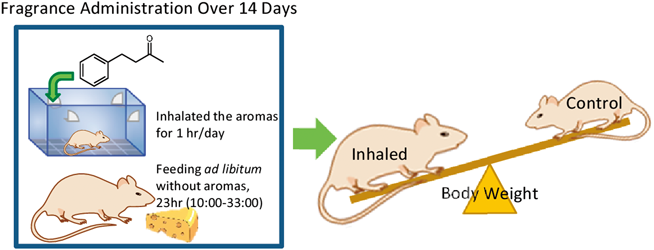 PDF形式でダウンロード (1060K) HTML形式で全画面表示
PDF形式でダウンロード (1060K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 799-806
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (1264K) HTML形式で全画面表示
PDF形式でダウンロード (1264K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 807-814
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/02/19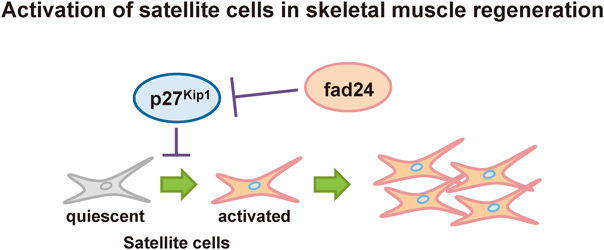 PDF形式でダウンロード (1737K) HTML形式で全画面表示
PDF形式でダウンロード (1737K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 815-822
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/02/19 PDF形式でダウンロード (2973K) HTML形式で全画面表示
PDF形式でダウンロード (2973K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 823-831
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (1094K) HTML形式で全画面表示
PDF形式でダウンロード (1094K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 832-838
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/02/09 PDF形式でダウンロード (4237K) HTML形式で全画面表示
PDF形式でダウンロード (4237K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 839-848
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/02/17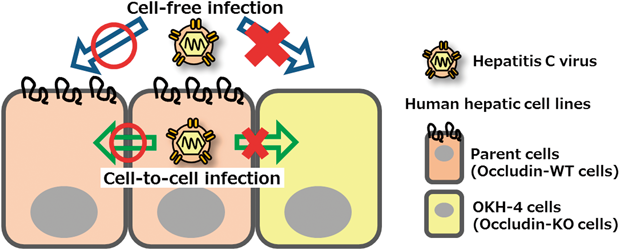 PDF形式でダウンロード (6098K) HTML形式で全画面表示
PDF形式でダウンロード (6098K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 849-855
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/03/04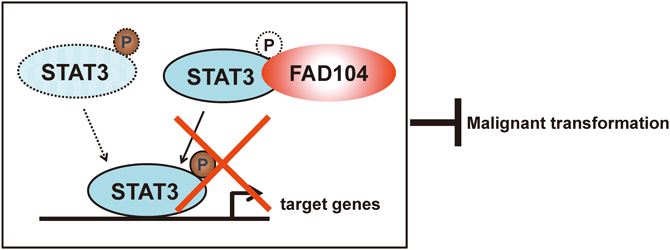 PDF形式でダウンロード (1272K) HTML形式で全画面表示
PDF形式でダウンロード (1272K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 856-862
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (827K) HTML形式で全画面表示
PDF形式でダウンロード (827K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 863-868
発行日: 2016/05/01
公開日: 2016/05/01
[早期公開] 公開日: 2016/03/04 PDF形式でダウンロード (680K) HTML形式で全画面表示
PDF形式でダウンロード (680K) HTML形式で全画面表示
Notes
-
2016 年 39 巻 5 号 p. 869-873
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (1080K) HTML形式で全画面表示
PDF形式でダウンロード (1080K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 874-878
発行日: 2016/05/01
公開日: 2016/05/01
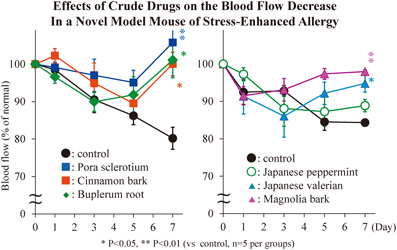 PDF形式でダウンロード (570K) HTML形式で全画面表示
PDF形式でダウンロード (570K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 879-882
発行日: 2016/05/01
公開日: 2016/05/01
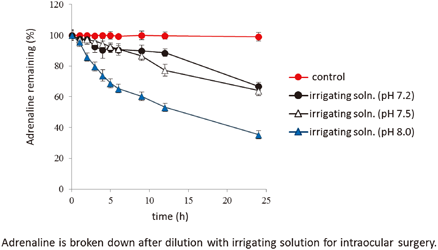 PDF形式でダウンロード (330K) HTML形式で全画面表示
PDF形式でダウンロード (330K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 883-886
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (378K) HTML形式で全画面表示
PDF形式でダウンロード (378K) HTML形式で全画面表示 -
2016 年 39 巻 5 号 p. 887-890
発行日: 2016/05/01
公開日: 2016/05/01
 PDF形式でダウンロード (587K) HTML形式で全画面表示
PDF形式でダウンロード (587K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|