
- |<
- <
- 1
- >
- >|
-
Risa Takayanagi, Takumi Uchida, Koji Kimura, Yasuhiko Yamada2018 年 41 巻 2 号 p. 153-157
発行日: 2018/02/01
公開日: 2018/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLGlucagon-like peptide-1 (GLP-1) receptor agonists (liraglutide, exenatide, lixisenatide) have recently been used as anti-diabetes drugs. We examined relationships of the binding occupancy of GLP-1 receptors (Φ) and their clinical efficacy after administration of GLP-1 receptor agonists. Next, by focusing on changes of GLP-1 concentration after administration of dipeptidyl peptidase-4 (DPP-4) inhibitors (vildagliptin, alogliptin, sitagliptin, linagliptin), we analyzed the relationship between Φ and clinical efficacy. Furthermore, using Φ as a common parameter, we compared the clinical efficacy elicited by GLP-1 receptor agonists and DPP-4 inhibitors using a theoretical analysis method. The present results showed that GLP-1 receptor agonists produced their clinical effect at a relatively low level of Φ (1.1–10.7%) at a usual dose. Furthermore, it was suggested that the drugs might achieve their full effect at an extraordinarily low level of Φ. It was also revealed that the Φ value of DPP-4 inhibitors (0.83–1.3%) was at the lower end or lower than that of GLP-1 receptor agonists at a usual dose. Accordingly, the predicted value for hemoglobin A1c (HbA1c) reduction after administration of the GLP-1 receptor agonists was higher than that of DPP-4 inhibitors. We clarified the differences between the therapeutic effects associated with GLP-1 receptor agonists and DPP-4 inhibitors theoretically. Together, the present findings provide a useful methodology for proper usage of GLP-1 receptor agonists and DPP-4 inhibitors.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (479K) HTML形式で全画面表示 -
Zhihua Yue, Jinhai Shi, Haona Li, Huiyi Li2018 年 41 巻 2 号 p. 158-162
発行日: 2018/02/01
公開日: 2018/02/01
[早期公開] 公開日: 2017/11/28 ジャーナル フリー HTML
ジャーナル フリー HTMLNonsteroidal anti-inflammatory drugs (NSAIDs) are likely to be used concomitantly with acyclovir or valacyclovir in clinical practice, but the study on the safety of such combinations was seldom reported. The objective of the study was to investigate reports of acute kidney injury (AKI) events associated with the concomitant use of oral acyclovir or valacyclovir with an NSAID by using the United States Food and Drug Administration (FDA) Adverse Event Reporting System (AERS) database between January 2004 and June 2012. The frequency of AKI events in patients while simultaneously taking either acyclovir or valacyclovir and an NSAID was compared using the Chi-square test. The effect of concomitant use of acyclovir or valacyclovir and individual NSAIDs on AKI was analyzed by the reporting odds ratio (ROR). The results showed that AKI was reported as the adverse event in 8.6% of the 10923 patients taking valacyclovir compared with 8.7% of the 2556 patients taking acyclovir (p=NS). However, AKI was significantly more frequently reported in patients simultaneously taking valacyclovir and an NSAID (19.4%) than in patients simultaneously taking acyclovir and an NSAID (10.5%) (p<0.01). The results also suggested that increased risk of AKI was likely associated with the concomitant use of valacyclovir and some NSAIDs such as loxoprofen, diclofenac, etodolac, ketorolac, piroxicam or lornoxicam. The case series from the AERS indicated that compared with acyclovir, valacyclovir is more likely to be affected by NSAIDs, and the concomitant use of valacyclovir with some NSAIDs might be associated with increased risk of AKI. The drug interactions with this specific combination of medications are worth exploring further.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (274K) HTML形式で全画面表示 -
Mitsuhiro Morita, Kotaro Yamada, Hideki Date, Kazue Hayakawa, Hidetomo ...2018 年 41 巻 2 号 p. 163-171
発行日: 2018/02/01
公開日: 2018/02/01
[早期公開] 公開日: 2017/11/27 ジャーナル フリー HTML
ジャーナル フリー HTMLWe explored the effects of chondroitin sulfate on knee osteoarthritis in a one-year, randomized, double-blind, dose-comparison study. Patients with painful, Kellgren–Lawrence grade 2–3, osteoarthritis of the knee were treated with oral chondroitin sulfate at a dose of either 260 mg/d (low-dose group, control group) or 1560 mg/d (high-dose group). Symptoms were evaluated by the Lequesne’s index and visual analog scale for pain. We made subgroup analyses according to background symptom severity (Lequesne’s index ≥8 or <8) in 73 patients. Serum level of cartilage oligomeric matrix protein and hyaluronic acid were also determined. In the subgroup with severe symptoms (Lequesne’s index ≥8), the chondroitin sulfate dose of 1560 mg/d improved pain faster after 6 and 9 months’ therapy. However, no dose-related effects were found on cartilage oligomeric matrix protein or hyaluronic acid levels. Chondroitin sulfate also had good tolerability. We conclude that chondroitin sulfate is useful for pain control in knee osteoarthritis.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (720K) HTML形式で全画面表示 -
Sol-Ji Kim, Ji-Hee Yeo, Seo-Yeon Yoon, Soon-Gu Kwon, Jang-Hern Lee, Al ...2018 年 41 巻 2 号 p. 172-181
発行日: 2018/02/01
公開日: 2018/02/01
[早期公開] 公開日: 2017/11/29 ジャーナル フリー HTML
ジャーナル フリー HTMLDespite the relatively high prevalence of migraine or headache, the pathophysiological mechanisms triggering headache-associated peripheral hypersensitivities, are unknown. Since nitric oxide (NO) is well known as a causative factor in the pathogenesis of migraine or migraine-associated hypersensitivities, a mouse model has been established using systemic administration of the NO donor, nitroglycerin (NTG). Here we tried to investigate the time course development of facial or hindpaw hypersensitivity after repetitive NTG injection. NTG (10 mg/kg) was administrated to mice every other day for nine days. Two hours post-injection, NTG produced acute mechanical and heat hypersensitivity in the hind paws. By contrast, cold allodynia, but not mechanical hypersensitivity, occurred in the facial region. Moreover, this hindpaws mechanical hypersensitivity and the facial cold allodynia was progressive and long-lasting. We subsequently examined whether the depletion of capsaicin-sensitive primary afferents (CSPAs) with resiniferatoxin (RTX, 0.02 mg/kg) altered these peripheral hypersensitivities in NTG-treated mice. RTX pretreatment did not affect the NTG-induced mechanical allodynia in the hind paws nor the cold allodynia in the facial region, but it did inhibit the development of hind paw heat hyperalgesia. Similarly, NTG injection produced significant hindpaw mechanical allodynia or facial cold allodynia, but not heat hyperalgesia in transient receptor potential type V1 (TRPV1) knockout mice. These findings demonstrate that different peripheral hypersensitivities develop in the face versus hindpaw regions in a mouse model of repetitive NTG-induced migraine, and that these hindpaw mechanical hypersensitivity and facial cold allodynia are not mediated by the activation of CSPAs.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2052K) HTML形式で全画面表示 -
Yasuyuki Fujimoto, Takashi Fujita, Nobuyuki Kuramoto, Mitsuru Kuwamura ...2018 年 41 巻 2 号 p. 182-189
発行日: 2018/02/01
公開日: 2018/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLInterleukin (IL)-19 is a member of the IL-10 family of interleukins and is an immuno-modulatory cytokine produced by the main macrophages. The gastrointestinal tissues of IL-19 knockout mice show exacerbated experimental colitis mediated by the innate immune system and T cells. There is an increasing focus on the interaction and relationship of IL-19 with the function of T cells. Contact hypersensitivity (CHS) is T cell-mediated cutaneous inflammation. Therefore, we asked whether IL-19 causes CHS. We investigated the immunological role of IL-19 in CHS induced by 1-fluoro-2,4-dinitrofluorobenzene as a hapten. IL-19 was highly expressed in skin exposed to the hapten, and ear swelling was increased in IL-19 knockout mice. The exacerbation of the CHS response in IL-19 knockout mice correlated with increased levels of IL-17 and IL-6, but no alterations were noted in the production of interferon (IFN)γ and IL-4 in the T cells of the lymph nodes. In addition to the effect on T cell response, IL-19 knockout mice increased production of inflammatory cytokines. These results show that IL-19 suppressed hapten-dependent skin inflammation in the elicitation phase of CHS.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (4361K) HTML形式で全画面表示 -
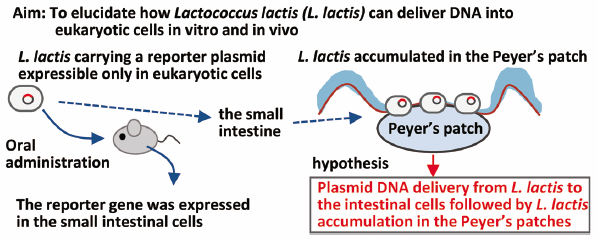
Ayumu Yano, Keita Takahashi, Yusuke Mori, Shiori Watanabe, Yuki Hanamu ...2018 年 41 巻 2 号 p. 190-197
発行日: 2018/02/01
公開日: 2018/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLApplication of food-grade Lactococcus lactis (L. lactis) as a safe delivery tool for DNA vaccines and therapeutic proteins has been well investigated. Although some studies showed that eukaryotic expression plasmids were transferred from L. lactis to enterocytes, the precise mechanism of the DNA transfer remains unknown. In this study, we generated an invasive L. lactis strain that expresses “murinized” Internalin A, an invasin of intracellular bacteria Listeria monocytogenes with two amino acid alterations for invasion into murine cells, and confirmed that this L. lactis strain delivered DNA in an invasin-dependent manner into a monolayer of epithelial cells polarized to mimic the gastrointestinal tract environment. Although invasive L. lactis inoculated orally can deliver DNA into enterocytes in the gastrointestinal tract of mice, the efficiency of DNA transfer was similar to that of non-invasive L. lactis strain, suggesting that the in vivo DNA transfer from L. lactis occurs invasin-independently. A ligated-intestinal loop assay, a method for a short-term culturing of the whole intestine filled with materials to evaluate the interaction of the materials with intestinal cells, demonstrated that both non-invasive and invasive L. lactis strains were present in the Peyer’s patches of the small intestine. On the other hand, few L. lactis was detected in the non-Peyer’s patch epithelial region. Thus, our observations lead us to speculate that DNA transfer from L. lactis occurs predominantly in the Peyer’s patches in an invasin-independent manner.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (3220K) HTML形式で全画面表示 -
Ying Yang, Xianglin Tang, Feiran Hao, Zengchun Ma, Yuguang Wang, Lili ...2018 年 41 巻 2 号 p. 198-207
発行日: 2018/02/01
公開日: 2018/02/01
[早期公開] 公開日: 2017/11/29 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録As a traditional herbal medicine, the fruits of Psoralea corylifolia L. (Fructus Psoraleae (FP)) have been widely used for the treatment of various skin diseases for hundred years. Recently, the emerging FP-induced toxic effects, especially hepatotoxicity, in clinic are getting the public’s attention. However, its exact toxic components and mechanisms underlying remain unclear. Bavachin, one of flavonoids in FP, has been documented as a hepatotoxic substance, and the present study aimed to determine the toxicity caused by bavachin and the possible toxic mechanisms involved using human hepatocellular carcinoma (HepG2) cells. Our results showed that bavachin could significantly inhibited cell proliferation and trigger the endoplasmic reticulum (ER) stress in a dose dependent manner. Downregulating ER stress using tauroursodeoxycholic acid (TUDCA) obvious attenuated bavachin-triggerd cell apoptosis. Then, small interfering RNA (siRNA) knock-down of Mitofusion2 (Mfn2) resulted in a remarkable aggravation of ER stress through the inhibition of the phosphorylation of protein kinase B (Akt). Additionally, suppression of reactive oxygen species (ROS) by ROS Scavenger (N-acetyl-l-cystein (NAC)) also reduced bavachin-induced ER stress. Taken together, our study demonstrated that bavachin-induced ER stress caused cell apoptosis by Mfn2-Akt pathway, and that ROS may participate upstream in this mechanism. Here, we not only provide a new understanding of ROS/Mfn2/Akt pathway in bavachin-induced cytotoxicity via the ER stress, but also identify a new specific intervention to prevent FP-induced hepatotoxicity in the future.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2629K) HTML形式で全画面表示 -
Takahiko Mitani, Kana Ota, Nobuya Inaba, Kunihiro Kishida, Hajime A. K ...2018 年 41 巻 2 号 p. 208-212
発行日: 2018/02/01
公開日: 2018/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLMume fruit, the Japanese apricot (Prunus mume SIEB. et ZUCC.), is popular in Japan and is mostly consumed in the pickled form called umeboshi. This fruit is known to have anti-microbial properties, but the principal constituents responsible for the antimicrobial properties have not yet been elucidated. We investigated the antimicrobial activities of the phenolic compounds in P. mume against enterobacteria. In this study, growth inhibitory activities were measured as an index of the antibacterial activities. The phenolic compounds were prepared from a byproduct of umeboshi called umesu or umezu (often translated as “mume vinegar”). Umesu or umezu phenolics (UP) contain approximately 20% phenolic compounds with p-coumaric acid as a standard and do not contain citric acid. We observed the inhibitory effects of UP against the growth of some enterobacteria, at a relatively high concentration (1250–5000 µg/mL). Alkali hydrolysates of UP (AHUP) exhibited similar antibacterial activities, but at much lower concentrations of 37.5–300 µg/mL. Since AHUP comprises hydroxycinnamic acids such as caffeic acid, p-coumaric acid, and ferulic acid, the antibacterial activities of each of these acids were examined. Our study shows that the phenolic compounds in P. mume other than citric acid contribute to its antimicrobial activity against enterobacteria in the digestive tract.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1844K) HTML形式で全画面表示 -
Hiroshi Kawai, Megumi Machida, Takuya Ishibashi, Naomi Kudo, Yoichi Ka ...2018 年 41 巻 2 号 p. 213-219
発行日: 2018/02/01
公開日: 2018/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLBiological rhythms are thought to be related to the pathogenesis and therapy of various diseases including depression. Here we investigated the influence of circadian rhythms on the antidepressant activity of the dual-action serotonin-noradrenaline reuptake inhibitor (SNRI) milnacipran. Rats administered milnacipran in the morning (8:00 a.m.; zeitgeber time [ZT]1) or in the evening (8:00 p.m.; ZT13) were analyzed in a forced swim test (FST). At ZT1, the rats’ immobility was reduced and the swimming was increased, whereas at ZT13, their climbing was increased. These results suggest that the serotonergic and noradrenergic systems are preferentially affected at ZT1 and ZT13, respectively by milnacipran. We analyzed the plasma and brain levels of milnacipran after administration, and there were no differences between ZT1 and ZT13. The circadian rhythm of monoamine neurotransmitters was analyzed in several brain regions. The serotonin turnover showed rhythms with a peak during ZT18–ZT22 in hippocampus. The noradrenaline turnover showed rhythms with a peak during ZT22–ZT2. There was a difference of approx. 4 h between the serotonergic and noradrenergic systems. This time difference might be one of the factors that affect the action of milnacipran and contribute to the dosing time-dependent behavioral pattern in the FST.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (549K) HTML形式で全画面表示 -
Ryoko Tomita, Kenichiro Todoroki, Tadashi Hayama, Hideyuki Yoshida, To ...2018 年 41 巻 2 号 p. 220-228
発行日: 2018/02/01
公開日: 2018/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Previously, we developed a method to evaluate states of cells treated with anticancer drugs via the comprehensive analysis of amino acids, termed amino acid metabolomics. In the present study, we evaluated the effects of the anticancer drugs, gemcitabine hydrochloride and pyrvinium pamoate, on the proliferation of a pancreatic cancer cell line (PANC-1) under hypoglycemic conditions using amino acid metabolomics. Intracellular and extracellular amino acid profiles of PANC-1 were determined by hydrophilic interaction chromatography-tandem mass spectrometry with simple pretreatment. Changes to the drugs’ anticancer effects resulting from glucose starvation conditions were presented in score plots obtained from principal component analyses. In particular, the analysis of intracellular amino acids was found to be the superior approach because the results allowed a clearer assessment of the cell state. Further, orthogonal partial least squares discriminant analysis was performed to search for amino acid candidates that discriminate with anticancer drug-treated PANC-1 cells. We identified several amino acids that might be able to distinguish the drug-treated group from the control group. These results might provide a better understanding of the mechanisms underlying cell responses such as drug resistance or austerity. The present study is the first to evaluate the efficacy of anticancer drugs under glucose starvation based on the analysis of the variation of extracellular and intracellular amino acid profiles in vitro.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1022K) HTML形式で全画面表示 -
Meng Lu, Xian Jiang, Liquan Tong, Feng Zhang, Lin Ma, Xuesong Dong, Xu ...2018 年 41 巻 2 号 p. 229-238
発行日: 2018/02/01
公開日: 2018/02/01
[早期公開] 公開日: 2017/11/30 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Maintaining a certain level of hydrogen sulfide (H2S) in ischemia–reperfusion (I/R) is essential for limiting injury to the liver. Exogenous H2S exerts protective effects against this injury, but the mechanisms remain unclear. Liver injury was induced in Wistar rats undergoing hepatic I/R for 30 min, followed by a 3-h reperfusion. Administration of GYY4137 (a slow-releasing H2S donor) significantly attenuated the severity of liver injury and was reflected by reduced inflammatory cytokine production and cell apoptosis, the levels of which were elevated by I/R, while DL-propargylglycine (PAG, an inhibitor of cystathionine γ-lyase [CSE]) aggravated liver injury. Delivery of GYY4137 significantly elevated the plasma levels of H2S and upregulated the expression of microRNA-21 (miR-21), leading to the activation of the Akt pathway, in rat livers subjected to I/R. To further investigate the protective mechanisms of H2S during liver I/R injury, we established a cell model of hypoxia/reoxygenation (H/R) by incubating Buffalo rat liver (BRL) cells under hypoxia for 4 h followed by normoxia for 10 h. The regulatory effect of miR-21 on the Akt pathway by downregulating phosphatase and tensin homolog (PTEN) was validated by luciferase assays. Incubation of sodium hydrosulfide (NaHS), an H2S donor, increased the expression of miR-21, attenuated the reduced cell viability and the increased apoptosis by H/R, in BRL cells. Anti-miR-21 abolished the protective effects of NaHS by inactivating the Akt pathway. In conclusion, the present results indicate the activation of the Akt pathway regulated by miR-21 participates in the protective effects of H2S against I/R-induced liver injury.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1851K) HTML形式で全画面表示 -
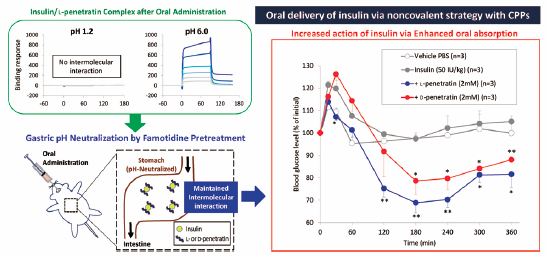
 Noriyasu Kamei, Chikako Shigei, Ryota Hasegawa, Mariko Takeda-Morishit ...2018 年 41 巻 2 号 p. 239-246
Noriyasu Kamei, Chikako Shigei, Ryota Hasegawa, Mariko Takeda-Morishit ...2018 年 41 巻 2 号 p. 239-246
発行日: 2018/02/01
公開日: 2018/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThis present study aimed to determine the optimal oral insulin delivery conditions that would maximize the utility of cell-penetrating peptides (CPPs) by using a noncovalent strategy. We first compared the effectiveness of two potential CPPs, penetratin and its analog PenetraMax, as absorption enhancers for insulin. The combined effect was evaluated under in vivo oral administration conditions. Both D-forms of CPPs were highly effective for increasing the oral absorption of insulin, and D-PenetraMax showed a more rapid onset of absorption enhancement effects compared with those of D-penetratin. However, synergistic absorption enhancement effects after combination treatment were not observed. Next, we tried a theoretical approach to establish optimized oral insulin delivery conditions. A surface plasmon resonance (SPR)-based analysis demonstrated that binding between insulin and penetratin (2 mM) might be saturated at 100–500 µM penetratin, while the bound concentration of penetratin could increase in accordance with an increased concentration of mixed insulin. To test this hypothesis, we investigated the effectiveness of different insulin doses in the gastric pH-neutralized mice. The results showed that the dissociation of noncovalent complexes of insulin and CPPs at the low gastric pH was prevented in these mice. Our findings clearly suggested that a noncovalent strategy with CPPs represents an effective approach for the L-form of CPP to increase the concentration of CPP-bound insulin to attain greater absorption of insulin, although this approach may not be appropriate for the D-form of CPP. Our findings will contribute to the development of oral dosage forms of insulin for noncovalent strategies involving CPP.
Graphical Abstract Fullsize Image抄録全体を表示Editor's pickBased on the binding characteristics, Kamei et al. attempted to increase the bound concentration of penetratin, cell-penetrating peptide (CPP), by increasing the concentration of mixed insulin, however the effect of L-penetratin on the oral absorption of insulin was not boosted by increasing the dose of insulin. The investigation in the gastric pH-neutralized mice showed that the dissociation of noncovalent complexes of insulin and CPPs at the low gastric pH was prevented in these mice, and clearly suggested that a noncovalent theoretical strategy with CPPs represents an effective approach for the L-form of CPP to attain greater absorption of insulin.
PDF形式でダウンロード (1483K) HTML形式で全画面表示 -
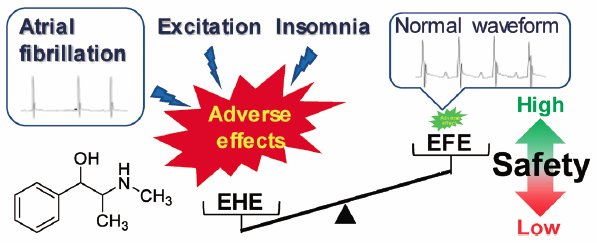
Hiroaki Takemoto, Jun Takahashi, Sumiko Hyuga, Hiroshi Odaguchi, Nahok ...2018 年 41 巻 2 号 p. 247-253
発行日: 2018/02/01
公開日: 2018/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Ephedrine alkaloids-free Ephedra Herb extract (EFE) has been developed to eliminate the adverse effects caused by ephedrine alkaloid-induced sympathetic hyperactivation. Previously, we reported that EFE possesses analgesic, anti-influenza, and cancer metastatic inhibitory effects at comparable levels to that of Ephedra Herb extract (EHE). However, it has not yet been demonstrated that EFE is free from the known side effects of EHE, such as excitation, insomnia, and arrhythmias. In this study, the incidence of these adverse effects was compared between mice administered EHE and those administered EFE. Increased locomotor activity in an open-field test, reduced immobility times in a forced swim test, and reduced sleep times in a pentobarbital-induced sleep test were observed in EHE-treated mice, when compared to the corresponding values in vehicle-treated mice. In contrast, EFE had no obvious effects in these tests. In electrocardiograms, atrial fibrillation (i.e., irregular heart rhythm, absence of P waves, and appearance of f waves) was observed in the EHE-treated mice. It was suggested that this atrial fibrillation was induced by stimulation of adrenaline β1 receptors, but not by hypokalemia. However, EFE did not affect cardiac electrophysiology. These results suggest that the abovementioned side effects are caused by ephedrine alkaloids in EHE, and that EFE is free from these adverse effects, such as excitation, insomnia, and arrhythmias. Thus, EFE is a promising new botanical drug with few adverse effects.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (877K) HTML形式で全画面表示 -
Takeo Yasu, Kenji Momo, Shunsuke Kobayashi, Seiichirou Kuroda, Arinobu ...2018 年 41 巻 2 号 p. 254-258
発行日: 2018/02/01
公開日: 2018/02/01
[早期公開] 公開日: 2017/12/06 ジャーナル フリー HTML
ジャーナル フリー HTMLPonatinib, a novel tyrosine kinase inhibitor marketed in 2016, is a key drug used for treating chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. This study aimed to develop a simple method for determining plasma ponatinib concentration. The analysis required extraction of a 400-µL sample of plasma and precipitation of proteins using an Oasis HLB cartridge. Ponatinib and bosutinib, which is used as an internal standard, were separated by HPLC using a mobile phase of acetonitrile : 0.037 mol/L KH2PO4 (pH 4.5) (39 : 61, v/v) on a Capcell Pack C18 MG II (25×4.6 mm) monitored at 250 nm, with a flow rate of 1.0 mL/min. This assay method was then used for determining plasma ponatinib concentration in a 42-year-old man treated with ponatinib at 15 mg/d. The calibration curve was found to be linear for the plasma concentration range of 5–250 ng/mL with a regression coefficient (r2) of 0.9999. The coefficients of intra-day and inter-day validation under these concentrations were 2.1–6.0 and 4.5–8.0%, respectively. The assay accuracy was −1.5–9.0%, and the recovery was greater than 86%. The plasma concentration of the patient at 2.5 and 3 h after 15 mg ponatinib administration was 43.6 and 49.3 ng/mL, respectively. This method of HPLC equipped with UV detection for determining plasma ponatinib concentration has several advantages, such as simplicity and applicability to routine therapeutic drug monitoring at hospital laboratories.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (524K) HTML形式で全画面表示 -
Jonghwan Jegal, No-June Park, Sang-a Park, Sim-Kyu Bong, Hyun Jegal, S ...2018 年 41 巻 2 号 p. 259-265
発行日: 2018/02/01
公開日: 2018/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLJuniperus chinensis, commonly Chinese juniper, has been used for treating inflammatory diseases. This study aimed to investigate anti-atopic dermatitis (AD) effects of standardized J. chinensis fruits extract on murine oxazolone- and 2,4-dinitrochlorobenzene (DNCB)-induced models of AD. Ear swelling, epidermis thickening, and eosinophils infiltration in the oxazolone-mediated dermatitis of BALB/c mice were significantly reduced upon topical application of J. chinensis fruits 95% EtOH extract (JCE). Besides, transdermal administration of JCE to SKH-1 hairless mice inhibited the development of DNCB-induced AD-like skin lesions by suppressing transepidermal water loss and improving skin hydration. Decreased total serum immunoglobulin E (IgE) and interleukin (IL)-4 levels could be observed in atopic dorsal skin samples of JCE-treated group. According to the phytochemical analysis, JCE was found to contain isoscutellarein-7-O-β-D-xyloside, cupressuflavone, and amentoflavone as main compounds. Therapeutic attempts with the J. chinensis fruits might be useful in the treatment of AD and related skin inflammatory diseases.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2420K) HTML形式で全画面表示 -
Yasushi Nishioka, Kazuki Tamai, Masanari Onda, Youhei Hiromori, Tomoki ...2018 年 41 巻 2 号 p. 266-271
発行日: 2018/02/01
公開日: 2018/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLCorn oil, sesame oil, and 10% ethanol in corn oil are commonly used as dosing vehicles in toxicology studies. Since these vegetable oils contain bioactive compounds, it is important for toxicology studies to characterize the toxicities of the dosing vehicles themselves. It has been recently proposed that the width of the genital tubercle (GT), the dorsal–ventral length (D–V length) of the GT, and urethral tube closure in mouse fetuses can be used as novel markers for monitoring sexual development in mice. However, how these parameters are influenced by the dosing vehicles themselves remains unclear. Therefore, we evaluated the effects of corn oil, sesame oil, and 10% ethanol in corn oil on GT width, D–V length, and GT morphology in ICR mice. Our results showed that all three vehicles influenced GT width and D–V length, but not GT morphology, suggesting that the effects of dosing vehicles themselves might need to be considered when GT width or D–V length is used as a parameter to evaluate the effects of chemicals on GT development.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (3784K) HTML形式で全画面表示 -
Mari Akagawa, Asami Mori, Kenji Sakamoto, Tsutomu Nakahara2018 年 41 巻 2 号 p. 272-276
発行日: 2018/02/01
公開日: 2018/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Methylglyoxal, a highly reactive dicarbonyl compound, is formed as a by-product of glycolysis and plays an important role in the pathogenesis of diabetic complications, including diabetic retinopathy. However, it remains to be determined how methylglyoxal affects the regulatory mechanisms of retinal blood flow. In this study, we examined the effects of methylglyoxal on β2-adrenoceptor-mediated vasodilatory mechanisms in rat retinal arterioles. The retinal vasodilator responses were assessed by measuring the diameter of retinal arterioles in the fundus images. Intravitreal injection of methylglyoxal significantly diminished the vasodilation of retinal arterioles induced by the β2-adrenoceptor agonist salbutamol. The vasodilator effect of BMS-191011, a large-conductance Ca2+-activated K+ (BKCa) channel opener, on retinal arterioles was also attenuated by methylglyoxal. In contrast, methylglyoxal had no significant effect on retinal vasodilator response to forskolin. Methylglyoxal attenuated retinal vasodilator response to salbutamol under blockade of BKCa channels with iberiotoxin, an inhibitor of the channels. These results suggest that methylglyoxal attenuates β2-adrenoceptor-mediated retinal vasodilation by impairing the coupling of the β2-adrenoceptor to the guanine nucleotide-binding protein (Gs protein) and the function of the BKCa channel. Increased methylglyoxal in the eyes may contribute to the impairment of regulatory mechanisms of retinal blood flow in patients with diabetic retinopathy.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (634K) HTML形式で全画面表示
-
Akitoshi Tatsumi, Sachiyo Inoue, Tsuneo Hamaguchi, Seigo Iwakawa2018 年 41 巻 2 号 p. 277-280
発行日: 2018/02/01
公開日: 2018/02/01
[早期公開] 公開日: 2017/11/27 ジャーナル フリー HTML
ジャーナル フリー HTMLHuman serum albumin (HSA) has two major ligand-binding sites, sites I and II, and hydrolyzes compounds at both sites. Although the hydrolytic interaction of ester-type drugs with other drugs by HSA has been reported, there are only a few studies concerning the effect of pharmaceutical excipients on the hydrolysis of ester-type drugs by HSA. In the present study, we investigated the effect of ethanol (2 vol%; 345 mM) on the hydrolysis of aspirin, p-nitrophenyl acetate, and olmesartan medoxomil, which are ester-type drugs, with 4 different lots of HSA preparations. The hydrolysis activities of HSA toward aspirin, p-nitrophenyl acetate, and olmesartan medoxomil were measured from the pseudo-first-order degradation rate constant (kobs) of salicylic acid, p-nitrophenol, and olmesartan, respectively, which are the HSA-hydrolyzed products. Ethanol inhibited hydrolysis of aspirin by HSA containing low levels of fatty acids, but not by fatty acid-free HSA. Ethanol inhibited hydrolysis of p-nitrophenyl acetate by both fatty acid-free HSA and HSA containing low levels of fatty acids. In contrast, the hydrolysis of olmesartan medoxomil by HSA was insignificantly inhibited by ethanol, but inhibited not only by warfarin and indomethacin but also by naproxen, which are site I binding drugs and a site II binding drug, respectively. These results suggest that the inhibitory action of ethanol on the hydrolysis of ester-type drugs by HSA differs between site I binding drugs and site II binding drugs.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (339K) HTML形式で全画面表示 -
Azjargal Enkhsaikhan, Akira Takahara, Yuji Nakamura, Ai Goto, Koki Chi ...2018 年 41 巻 2 号 p. 281-284
発行日: 2018/02/01
公開日: 2018/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLA beverage made of red wine vinegar and grape juice (Yamanashi-no-megumi™) was developed as a supplemental fluid containing polyphenols, which has been clinically shown to enhance the colonic transit. In this study, we assessed the mechanism of its prokinetic action by analyzing the effects on both the colonic phosphodiesterase activity of rats (n=4) and the isolated colonic strip preparation of guinea pigs (n=4). The 7% (v/v) solution of the beverage significantly decreased the phosphodiesterase activity by 9% (n=4). The beverage in concentrations of 0.7, 2.1 and 7% (v/v) relaxed the colonic strips pre-contracted by 1 µmol/L of carbachol in a concentration-related manner with 50, 58 and 79%, each response of which was diminished to 11, 19 and 46%, respectively in the presence of 100 µmol/L of L-nitro-arginine methyl ester. These results obtained by biochemical, functional and pharmacological analyses suggest that the beverage could relax the colon through both cAMP-associated and nitric oxide-dependent pathways, which may partly explain clinically observed prokinetic effect of the beverage.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (454K) HTML形式で全画面表示
-
2018 年 41 巻 2 号 p. 285
発行日: 2018/02/01
公開日: 2018/02/01
ジャーナル フリー HTMLPDF形式でダウンロード (92K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|