
40 巻, 10 号
選択された号の論文の31件中1~31を表示しています
- |<
- <
- 1
- >
- >|
Reviews
-
2017 年 40 巻 10 号 p. 1605-1615
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (3734K) HTML形式で全画面表示
PDF形式でダウンロード (3734K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1616-1624
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (784K) HTML形式で全画面表示
PDF形式でダウンロード (784K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1625-1629
発行日: 2017/10/01
公開日: 2017/10/01
[早期公開] 公開日: 2017/08/01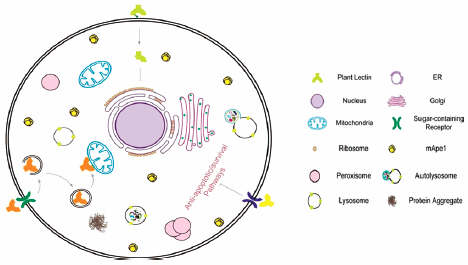 PDF形式でダウンロード (710K) HTML形式で全画面表示
PDF形式でダウンロード (710K) HTML形式で全画面表示
Regular Articles
-
2017 年 40 巻 10 号 p. 1630-1637
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (3340K) HTML形式で全画面表示
PDF形式でダウンロード (3340K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1638-1645
発行日: 2017/10/01
公開日: 2017/10/01
[早期公開] 公開日: 2017/07/26 PDF形式でダウンロード (708K) HTML形式で全画面表示
PDF形式でダウンロード (708K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1646-1653
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (10841K) HTML形式で全画面表示
PDF形式でダウンロード (10841K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1654-1660
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (863K) HTML形式で全画面表示
PDF形式でダウンロード (863K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1661-1668
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (1910K) HTML形式で全画面表示
PDF形式でダウンロード (1910K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1669-1677
発行日: 2017/10/01
公開日: 2017/10/01
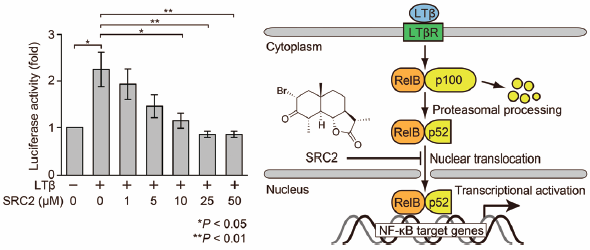 PDF形式でダウンロード (2844K) HTML形式で全画面表示
PDF形式でダウンロード (2844K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1678-1685
発行日: 2017/10/01
公開日: 2017/10/01
[早期公開] 公開日: 2017/08/11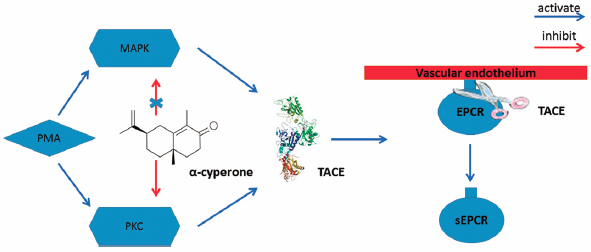 PDF形式でダウンロード (1577K) HTML形式で全画面表示
PDF形式でダウンロード (1577K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1686-1692
発行日: 2017/10/01
公開日: 2017/10/01
[早期公開] 公開日: 2017/07/21 PDF形式でダウンロード (4080K) HTML形式で全画面表示
PDF形式でダウンロード (4080K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1693-1699
発行日: 2017/10/01
公開日: 2017/10/01
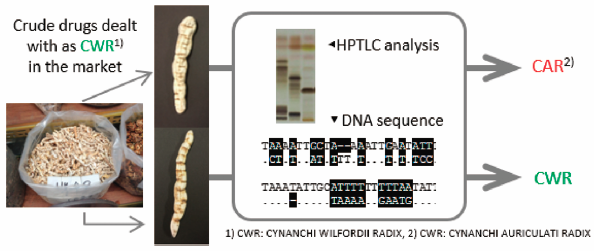 PDF形式でダウンロード (4762K) HTML形式で全画面表示
PDF形式でダウンロード (4762K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1700-1705
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (409K) HTML形式で全画面表示
PDF形式でダウンロード (409K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1706-1715
発行日: 2017/10/01
公開日: 2017/10/01
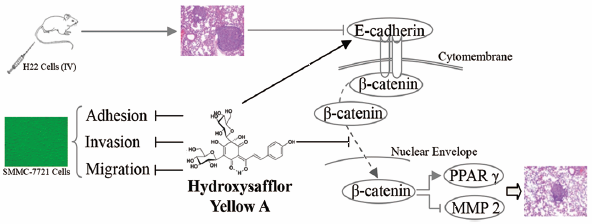 PDF形式でダウンロード (5238K) HTML形式で全画面表示
PDF形式でダウンロード (5238K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1716-1723
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (6287K) HTML形式で全画面表示
PDF形式でダウンロード (6287K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1724-1729
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (2057K) HTML形式で全画面表示
PDF形式でダウンロード (2057K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1730-1738
発行日: 2017/10/01
公開日: 2017/10/01
[早期公開] 公開日: 2017/08/04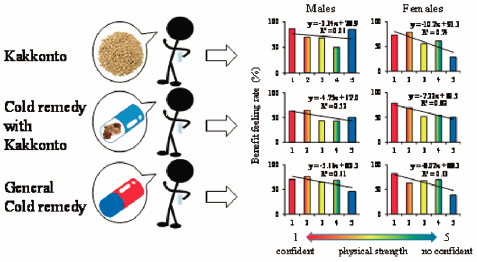 PDF形式でダウンロード (513K) HTML形式で全画面表示
PDF形式でダウンロード (513K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1739-1746
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (1161K) HTML形式で全画面表示
PDF形式でダウンロード (1161K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1747-1753
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (2256K) HTML形式で全画面表示
PDF形式でダウンロード (2256K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1754-1758
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (569K) HTML形式で全画面表示
PDF形式でダウンロード (569K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1759-1766
発行日: 2017/10/01
公開日: 2017/10/01
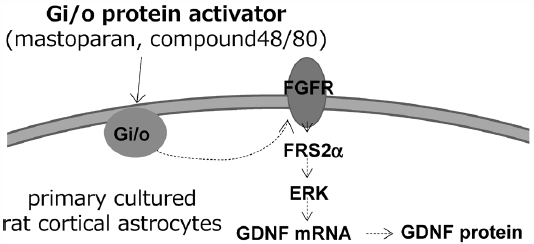 PDF形式でダウンロード (707K) HTML形式で全画面表示
PDF形式でダウンロード (707K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1767-1774
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (2244K) HTML形式で全画面表示
PDF形式でダウンロード (2244K) HTML形式で全画面表示
Notes
-
2017 年 40 巻 10 号 p. 1775-1778
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (320K) HTML形式で全画面表示
PDF形式でダウンロード (320K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1779-1783
発行日: 2017/10/01
公開日: 2017/10/01
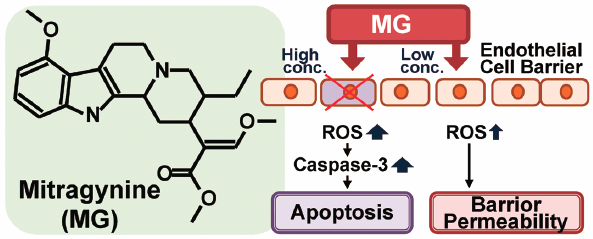 PDF形式でダウンロード (679K) HTML形式で全画面表示
PDF形式でダウンロード (679K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1784-1788
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (572K) HTML形式で全画面表示
PDF形式でダウンロード (572K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1789-1795
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (511K) HTML形式で全画面表示
PDF形式でダウンロード (511K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1796-1800
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (902K) HTML形式で全画面表示
PDF形式でダウンロード (902K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1801-1805
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (789K) HTML形式で全画面表示
PDF形式でダウンロード (789K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1806-1812
発行日: 2017/10/01
公開日: 2017/10/01
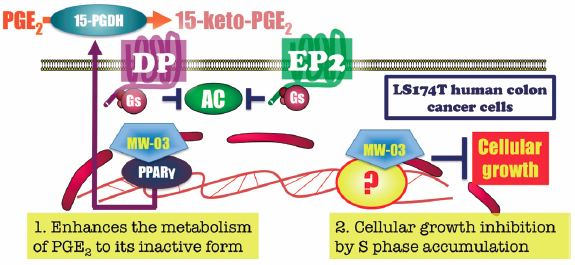 PDF形式でダウンロード (1718K) HTML形式で全画面表示
PDF形式でダウンロード (1718K) HTML形式で全画面表示 -
2017 年 40 巻 10 号 p. 1813-1817
発行日: 2017/10/01
公開日: 2017/10/01
 PDF形式でダウンロード (588K) HTML形式で全画面表示
PDF形式でダウンロード (588K) HTML形式で全画面表示
Errata
-
2017 年 40 巻 10 号 p. 1818
発行日: 2017/10/01
公開日: 2017/10/01
PDF形式でダウンロード (81K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|