
40 巻, 12 号
選択された号の論文の28件中1~28を表示しています
- |<
- <
- 1
- >
- >|
Reviews
-
2017 年 40 巻 12 号 p. 2015-2023
発行日: 2017/12/01
公開日: 2017/12/01
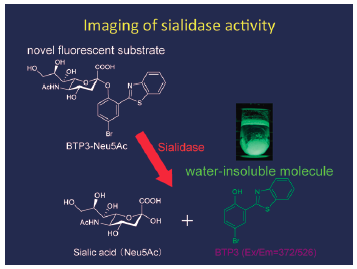 PDF形式でダウンロード (14112K) HTML形式で全画面表示
PDF形式でダウンロード (14112K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2024-2037
発行日: 2017/12/01
公開日: 2017/12/01
[早期公開] 公開日: 2017/10/07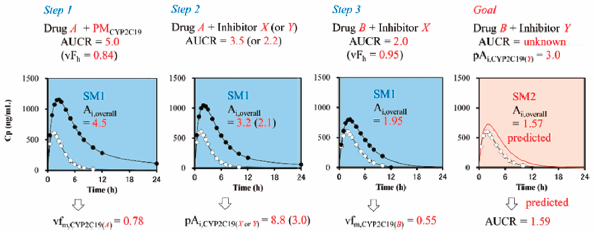 PDF形式でダウンロード (2885K) HTML形式で全画面表示
PDF形式でダウンロード (2885K) HTML形式で全画面表示
Current Topics - Recent Progress in the Study of Vasoactive Modulators in Metabolic and Cardiovascular Diseases
-
2017 年 40 巻 12 号 p. 2038
発行日: 2017/12/01
公開日: 2017/12/01
PDF形式でダウンロード (153K) HTML形式で全画面表示
Current Topics : Reviews
-
2017 年 40 巻 12 号 p. 2039-2044
発行日: 2017/12/01
公開日: 2017/12/01
 PDF形式でダウンロード (802K) HTML形式で全画面表示
PDF形式でダウンロード (802K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2045-2049
発行日: 2017/12/01
公開日: 2017/12/01
 PDF形式でダウンロード (6320K) HTML形式で全画面表示
PDF形式でダウンロード (6320K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2050-2060
発行日: 2017/12/01
公開日: 2017/12/01
 PDF形式でダウンロード (891K) HTML形式で全画面表示
PDF形式でダウンロード (891K) HTML形式で全画面表示
Current Topics : Regular Article
-
2017 年 40 巻 12 号 p. 2061-2067
発行日: 2017/12/01
公開日: 2017/12/01
 PDF形式でダウンロード (990K) HTML形式で全画面表示
PDF形式でダウンロード (990K) HTML形式で全画面表示
Regular Articles
-
2017 年 40 巻 12 号 p. 2068-2074
発行日: 2017/12/01
公開日: 2017/12/01
[早期公開] 公開日: 2017/09/22 PDF形式でダウンロード (1999K) HTML形式で全画面表示
PDF形式でダウンロード (1999K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2075-2080
発行日: 2017/12/01
公開日: 2017/12/01
 PDF形式でダウンロード (1251K) HTML形式で全画面表示
PDF形式でダウンロード (1251K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2081-2087
発行日: 2017/12/01
公開日: 2017/12/01
[早期公開] 公開日: 2017/09/23 PDF形式でダウンロード (1024K) HTML形式で全画面表示
PDF形式でダウンロード (1024K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2088-2095
発行日: 2017/12/01
公開日: 2017/12/01
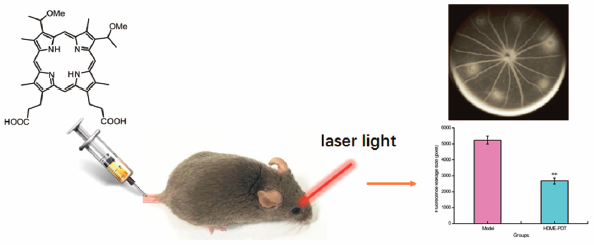 PDF形式でダウンロード (5446K) HTML形式で全画面表示
PDF形式でダウンロード (5446K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2096-2104
発行日: 2017/12/01
公開日: 2017/12/01
 PDF形式でダウンロード (1490K) HTML形式で全画面表示
PDF形式でダウンロード (1490K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2105-2109
発行日: 2017/12/01
公開日: 2017/12/01
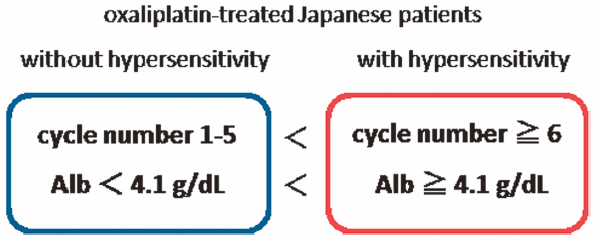 PDF形式でダウンロード (471K) HTML形式で全画面表示
PDF形式でダウンロード (471K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2110-2116
発行日: 2017/12/01
公開日: 2017/12/01
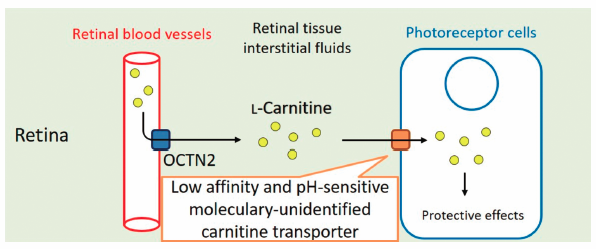 PDF形式でダウンロード (929K) HTML形式で全画面表示
PDF形式でダウンロード (929K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2117-2124
発行日: 2017/12/01
公開日: 2017/12/01
[早期公開] 公開日: 2017/09/30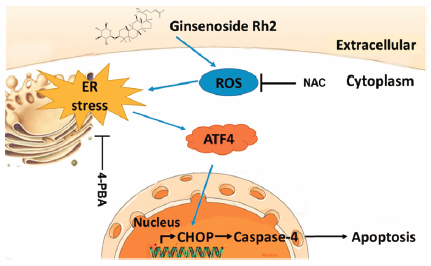 PDF形式でダウンロード (2916K) HTML形式で全画面表示
PDF形式でダウンロード (2916K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2125-2133
発行日: 2017/12/01
公開日: 2017/12/01
[早期公開] 公開日: 2017/09/22 PDF形式でダウンロード (2544K) HTML形式で全画面表示
PDF形式でダウンロード (2544K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2134-2139
発行日: 2017/12/01
公開日: 2017/12/01
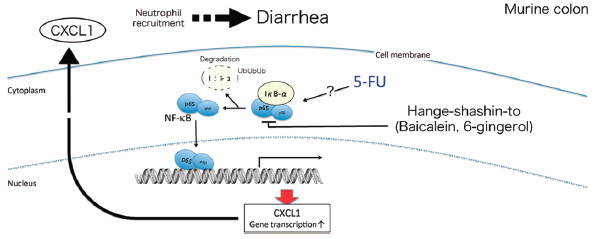 PDF形式でダウンロード (714K) HTML形式で全画面表示
PDF形式でダウンロード (714K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2140-2145
発行日: 2017/12/01
公開日: 2017/12/01
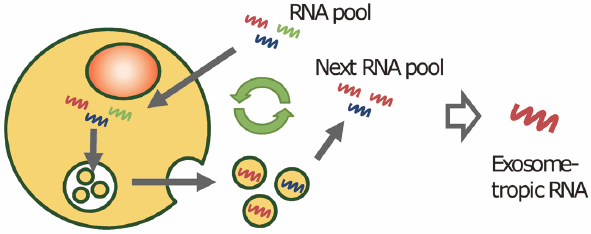 PDF形式でダウンロード (524K) HTML形式で全画面表示
PDF形式でダウンロード (524K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2146-2152
発行日: 2017/12/01
公開日: 2017/12/01
[早期公開] 公開日: 2017/10/04 PDF形式でダウンロード (3918K) HTML形式で全画面表示
PDF形式でダウンロード (3918K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2153-2157
発行日: 2017/12/01
公開日: 2017/12/01
[早期公開] 公開日: 2017/09/29 PDF形式でダウンロード (423K) HTML形式で全画面表示
PDF形式でダウンロード (423K) HTML形式で全画面表示 -
 2017 年 40 巻 12 号 p. 2158-2165
2017 年 40 巻 12 号 p. 2158-2165
発行日: 2017/12/01
公開日: 2017/12/01
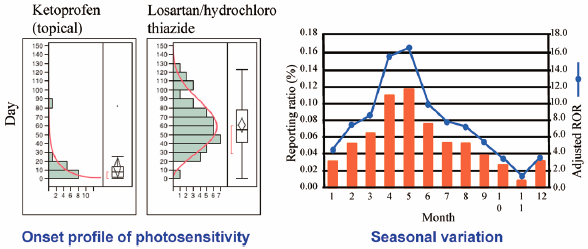
-
2017 年 40 巻 12 号 p. 2166-2174
発行日: 2017/12/01
公開日: 2017/12/01
[早期公開] 公開日: 2017/09/30 PDF形式でダウンロード (1082K) HTML形式で全画面表示
PDF形式でダウンロード (1082K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2175-2182
発行日: 2017/12/01
公開日: 2017/12/01
 PDF形式でダウンロード (661K) HTML形式で全画面表示
PDF形式でダウンロード (661K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2183-2190
発行日: 2017/12/01
公開日: 2017/12/01
 PDF形式でダウンロード (15544K) HTML形式で全画面表示
PDF形式でダウンロード (15544K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2191-2198
発行日: 2017/12/01
公開日: 2017/12/01
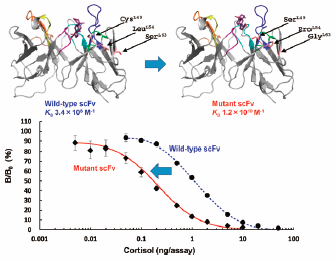 PDF形式でダウンロード (1940K) HTML形式で全画面表示
PDF形式でダウンロード (1940K) HTML形式で全画面表示
Notes
-
2017 年 40 巻 12 号 p. 2199-2204
発行日: 2017/12/01
公開日: 2017/12/01
[早期公開] 公開日: 2017/09/28 PDF形式でダウンロード (282K) HTML形式で全画面表示
PDF形式でダウンロード (282K) HTML形式で全画面表示 -
2017 年 40 巻 12 号 p. 2205-2211
発行日: 2017/12/01
公開日: 2017/12/01
 PDF形式でダウンロード (921K) HTML形式で全画面表示
PDF形式でダウンロード (921K) HTML形式で全画面表示
Errata
-
2017 年 40 巻 12 号 p. 2212
発行日: 2017/12/01
公開日: 2017/12/01
PDF形式でダウンロード (147K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|